


Содержание
Перейти к:
https://doi.org/10.24884/1561-6274-2018-22-2-74-80
Перейти к:
Болезнь Дента – Х-сцепленная проксимальная тубулопатия, характеризующаяся низкомолекулярной протеинурией, гиперкальциурией, нефрокальцинозом и прогрессированием в хроническую почечную недостаточность. Заболевание обусловлено мутациями в генах CLCN5 или OCRL и проявляется у лиц мужского пола, в то время как женщины являются бессимптомными носителями. ЦЕЛЬ: исследование фенотипа и генотипа матерей мальчиков с болезнью Дента для исключения бессимптомного носительства мутаций генов CLCN5 и OCRL, ответственных за развитие Х-сцепленной тубулопатии. ПАЦИЕНТЫ И МЕТОДЫ. Проведено клиническое и молекулярно-генетическое обследование 9 матерей 10 мальчиков с болезнью Дента из 8 неродственных семей. Прямое секвенирование по Сэнгеру генов CLCN5 и OCRL выполнено у всех пациентов и их мам. РЕЗУЛЬТАТЫ. Статус носителей болезни Дента 1-го (n=7) и 2-го (n=2) типов был подтвержден у всех обследованных мам пробандов. Превалирующими клиническими проявлениями болезни Дента у мам пациентов являлись снижение реабсорбции фосфатов, гипофосфатемия, медуллярный нефрокальциноз и прогрессирование в хроническую болезнь почек 2 стадии. Реже наблюдались низкомолекулярная протеинурия и гиперкальциурия. Полный фенотип болезни Дента выявлен у 2 женщин – двоюродных сестер, носителей болезни Дента 1-го типа. ЗАКЛЮЧЕНИЕ. У мам мальчиков с болезнью Дента установлена фенотипическая вариабельность клинических проявлений Х-сцепленной тубулопатии, что может быть следствием транслокации или инактивации Х-хромосомы. Полученные данные свидетельствуют о необходимости диагностического обследования матерей мальчиков с болезнью Дента с целью определения фенотипа и своевременной профилактики прогрессирования заболевания в хроническую почечную недостаточность.
Приходина Л.С., Папиж С.В., Баширова З.Р., Людвиг М. ЯВЛЯЮТСЯ ЛИ МАМЫ МАЛЬЧИКОВ С БОЛЕЗНЬЮ ДЕНТА БЕССИМПТОМНЫМИ НОСИТЕЛЯМИ Х-СЦЕПЛЕННОЙ ТУБУЛОПАТИИ? Нефрология. 2018;22(2):74-80. https://doi.org/10.24884/1561-6274-2018-22-2-74-80
Prikhodina L.S., Papizh S.V., Bashirova Z.R., Ludwig M. ARE MOTHERS OF BOYS WITH DENT’S DISEASE ASYMPTOMATIC CARRIERS FOR X-LINKED TUBULAR DISORDER? Nephrology (Saint-Petersburg). 2018;22(2):74-80. (In Russ.) https://doi.org/10.24884/1561-6274-2018-22-2-74-80
Болезнь Дента - Х-сцепленная проксимальная тубулопатия, характеризующаяся низкомолекулярной протеинурией, гиперкальциурией, нефрокальцинозом и прогрессированием в хроническую почечную недостаточность (ХПН).
Впервые клиническое описание заболевания представлено C.E. Dent и M. Friedman в 1964 году [1]. Распространенность болезни Дента в настоящее время остается неизвестной, что, вероятно, связано с редкой диагностикой данного заболевания. Болезнь Дента представляет собой редкое заболевание, около 250 семей описано в литературе в настоящее время.
В 60% случаев заболевание обусловлено мутациями в гене CLCN5 (OMIM #300009), локализованном на хромосоме Xp11, ответственного за развитие болезни Дента 1-го типа [2, 3]. У 15% пациентов идентифицированы мутации в гене OCRL (OMIM #300555), локализованном на хромосоме Xq26.1, приводящие к болезни Дента 2-го типа [4]. У 25% пациентов с клиническими признаками болезни Дента не выявлены мутации в генах CLCN5 или OCRL, что, вероятно, связано с пока неизвестными генами-кандидатами заболевания [5].
Клинические проявления болезни Дента 1-го типа в детском возрасте у большинства мальчиков немногочисленны. Нередко первым признаком заболевания является случайно обнаруженная протеинурия 0,5 - 1 г/л, которая при обследовании является, преимущественно, низкомолекулярной и наблюдается у 100% пациентов [6]. Нефротическая протеинурия описана у 20-44% мальчиков с болезнью Дента 1-го типа [7, 8]. Гиперкальциурия наблюдается у 70-95% лиц мужского пола, нефрокальциноз отмечается в 75% случаев [6, 7]. Более чем у трети пациентов с болезнью Дента выявляются гипофосфатемия со снижением тубулярной реабсорбции фосфатов, уролитиаз, аминоациду- рия, рахитические деформации костей [9, 10].
Болезнь Дента 2-го типа нередко рассматривается как мягкий вариант синдрома Лоу и характеризуется сходным фенотипом с болезнью Дента 1-го типа, но в ряде случаев с невыраженными экстраренальными проявлениями в виде мышечной гипотонии, снижении IQ. У пациентов с болезнью Дента 2-го типа часто выявляются повышенный уровень в крови ферментов мышечной ткани лак- татдегидрогеназы и креатинфосфокиназы [11].
У большинства пациентов мужского пола с болезнью Дента отмечается медленно прогрессирующее снижение функций почек с формированием терминальной ХПН у 30-80% мужчин в возрасте 30-50 лет [11]. Снижение функций почек до ХБП 2 стадии в детском возрасте отмечено у 45% мальчиков с болезнью Дента 1-го типа [7].
В основе патогенеза болезни Дента лежит генетическое детерминированное нарушение транспорта в клетках проксимальных канальцев, сопровождаемое потерей мегалина и кубилина в апикальной щеточной каемке, дефектом апикального эндоцитоза, что клинически проявляется в виде низкомолекулярной протеинурии и развитием синдрома Фанкони [12]. Дисбаланс между различными звеньями патогенеза заболевания может приводить к широкой вариабельности клинических проявлений болезни Дента даже среди членов одной семьи при отсутствии установленных генотип-фенотипических ассоциаций [9, 10].
Болезнь Дента 1-го и 2-го типов клинически проявляется у лиц мужского пола, а женщины являются облигатными бессимптомными носителями заболевания в соответствии с Х-сцепленным типом наследования патологии [9, 13].
В ранее проведенных исследованиях продемонстрировано, что у женщин - гетерозиготных носителей болезни Дента не выявлено нефрокальциноза и/или уролитиаза, нарушения реабсорбции фосфатов, снижения функционального состояния почек [13].
В немногочисленных исследованиях у девочек - носителей мутаций в генах CLCN5 или OCRL установлен мягкий фенотип болезни Дента в виде низкомолекулярной протеинурии у 60-90%, гиперкальциурии - у 30%, редким выявлением нефрокальциноза и прогрессированием в ХПН [14, 15]. Появление клинических симптомов болезни Дента у лиц женского пола с мутациями в генах CLCN5 или OCRL может быть обусловлено инактивацией Х-хромосомы [13].
До настоящего времени остается неизученным вопрос наличия и выраженности клинических проявлений болезни Дента у женщин - носителей мутаций генов CLCN5 и OCRL и целесообразности их обследования для своевременного проведения терапевтических мероприятий.
Целью настоящего исследования являлось исследование фенотипа и генотипа матерей мальчиков с болезнью Дента для исключения/подтверждения бессимптомного носительства мутаций генов CLCN5 и OCRL, ответственных за развитие Х-сцепленной тубулопатии.
Проведено клиническое и молекулярно генетическое обследование 9 мам 10 мальчиков с болезнью Дента, наблюдавшихся в отделе наследственных и приобретенных болезней почек НИКИ педиатрии им. акад. Ю.Е. Вельтищева ФГБОУ ВО РНИМУ им. Н.И. Пирогова.
Медиана возраста мам пробандов на момент обследования составляла 35 (ИКР: 32-39) лет. Болезнь Дента у мальчиков клинически диагностирована на основании выявленной низкомолекулярной протеинурии в комбинации с гиперкальциурией, гипофосфатемией и нефрокальцинозом [16].
Низкомолекулярная протеинурия определялась при повышении экскреции β2-микроглобулина с мочой >300 нг/мл. Гиперкальциурия устанавливалась при повышении референсных значений экскреции кальция с мочой более 6,25 ммоль/сут в соответствии с возрастом и полом [17].
Гиперфосфатурия оценивалась при превышении 95-го перцентиля референсных значений экскреции фосфора в моче в соответствии с возрастом и полом [18]. Гипофосфатемия определялась при снижении уровня фосфора в крови менее 1,3 ммоль/л. Тубулярная реабсорбция фосфатов (ТРФ) рассчитывалась по формуле [19]:
Снижение ТРФ оценивалось при снижении расчетного параметра менее 85%. Расчет коэффициента максимальной ТРФ по отношению к рСКФ (ТмРФ/рСКФ) осуществляли по формулам [19]:
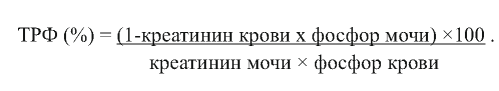
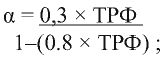
Снижение ТмР/рСКФ оценивалось при ТмРФ/ рСКФ <0,90 ммоль/л в соответствии с референс- ными значениями по полу и возрасту [19, 20].
Выраженность медуллярного нефрокальцино- за определяли при УЗИ в зависимости от степени повышения эхогенности медуллярных пирамид почек: 1 степень - фокальное повышение эхогенности пирамид, в том числе ободки вокруг пирамид; 2 степень - распространенное повышение эхогенности пирамид без акустической тени; 3 степень - выраженное гомогенное распространенное повышение эхогенности пирамид с акустической тенью [21]. Полиурия определялась как суточный объем выделенной мочи более 3 л/1,73 м2/сут.
Функциональное состояние почек у матерей пробандов оценивалось с определением расчетной скорости клубочковой фильтрации (рСКФ) по формуле CKD-EPI [22], у мальчиков с болезнью Дента - по формуле Шварца [23]. Последующая оценка функций почек по стадиям хронической болезни почек (ХБП) у женщин-носителей и их сыновей определялась в соответствии с классификацией K/DOQI [24].
Молекулярно-генетическое исследование проводилось с использованием геномной ДНК, выделенной из лейкоцитов крови всех матерей пробандов и их сыновей методом полимеразно-цепной реакции (ПЦР), амплификации и последующего автоматического секвенирования всех кодирующих и экзон-интронных регионов генов CLCN5 и OCRL с использованием ранее описанных праймеров и условий ПЦР [3, 25].
Характеристики идентифицированных мутаций в генах CLCN5 и OCRL представлены в соответствии с данными генетических баз: Online Mende- lian Inheritance in Man (OMIM) (https://www.omim.org), NCBI 1000 Genomes Browser (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes) и Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php).
Статистический анализ полученных данных проводили по общепринятым методикам вариационной статистики с использованием программы SPSS for Windows 17.0 (IBM Inc., США). Выбор критериев проверки гипотез проводился в зависимости от типа распределения. В случае отличия распределения от нормального применяли методы непараметрической статистики, для нормально распределенных данных в ходе их описания использовали среднее и среднеквадратичное отклонение (М±?). Результаты исследования представлены как медиана и интерквартильный размах (ИКР) (25-й; 75-й перцентили). Статистическую значимость различий двух средних определяли при помощи критерия Манна-Уитни, для оценки силы связи между изучаемыми переменными - коэффициент ранговой корреляции Спирмена. Нулевую статистическую гипотезу об отсутствии различий и связей отвергали при p<0,05.
Медиана возраста включенных в исследование мам мальчиков с болезнью Дента составляла 35 (ИКР: 32-39) лет.
Статус носителей болезни Дента 1-го (n=7) и 2-го (n=2) типов был подтвержден у всех обследованных мам пациентов. Проведенные молекулярногенетические исследования позволили идентифицировать патогенные гетерозиготные мутации в гене
CLCN5: с.1909С>Т (p.Ag637*) (n=2), с.731С>Т (p.Ser244Leu) (n=2), с.211С>Т (p.Arg707Ter) (n=1), с.842С>Т (p.Ser281Leu) (n=1), c.206-2A>G (n=1) и в гене OCRL (n=2): с.316А>Т (p.Lys106*) и с.1497С>Т (p.Arg493Trp) (табл. 1). В 5 (55,6%) случаях выявленные мутации ранее не описаны, включая в гене CLCN5 (n=3) и в гене OCRL (n=1).
Незначительная протеинурия, преимущественно низкомолекулярная в виде повышенной экскреции β2-микроглобулина, и гиперкальциурия выявлены у 2 (22,2%) мам мальчиков с болезнью Дента (табл. 2). Мочевого синдрома не отмечалось у 7 (77,8%) женщин - носителей данной патологии. Сниженная ТРФ определена у 6 (66,7%) обследованных мам пробандов с болезнью Дента, со сниженным коэффициентом ТмРФ/рСКФ у 5 (55,6%) женщин - носителей заболевания. Гипофосфатемия обнаружена у 7 (77,8%) обследованных женщин. Медуллярный нефрокальциноз 1 степени выявлен при УЗИ почек у 6 (66,7%) мам пробандов с болезнью Дента. Гиперфосфатурия и полиурия не выявлены ни у одной из обследованных женщин - облигатных носителей мутаций в генах CLCN5 и OCRL.
Таблица 1
Молекулярно-генетические характеристики мам мальчиков с болезнью Дента (n=9)
№ п/п | Носитель болезни Дента, типы | Ген | Экзон | Положен и е в ДНК | минокислотная амена | NCBI 1000 Genomes Browser | Тип мутации |
|---|---|---|---|---|---|---|---|
1 | 1-й | CLCN5 | 10 | c.1909C>T | p.Arg637* | rs151340626 | Нонсенс |
2§ | 1-й | CLCN5 | 8 | c.842C>T | p.Ser281Leu | новая | Миссенс |
3 | 1-й | CLCN5 | 10 | c.1909C>T | p.Arg637* | rs151340626 | Нонсенс |
4¥ | 1-й | CLCN5 | 7 | c.731 C>T | p.Ser244Leu | rs151340626 | Миссенс |
5¥ | 1-й | CLCN5 | 7 | c.731 C>T | p.Ser244Leu | rs151340626 | Миссенс |
6 | 1-й | CLCN5 | 4 | c.206-2A>G | - | новая | Сайта cплайсинга |
7 | 2-й | OCRL | 5 | c.316A>T | p. Lys 106* | новая | Нонсенс |
8 | 1-й | CLCN5 | 13 | c.211 C>T | p.Arg707Ter | новая | Миссенс |
9 | 2-й | OCRL | 15 | c. 1497C>T | p.Arg493Trp | новая | Миссенс |
Примечание. Здесь и в табл. 2: § мама 2-х сыновей с болезнью Дента; ¥ двоюродные сестры.
Таблица 2
Клинические характеристики мам мальчиков с болезнью Дента (n=9)
№ п/п | Возраст, годы | β2-микро- глобулин мочи, нг/мл | Кальций очи, ммоль/ сут | Фосфор крови, ммоль/л | Креатинин крови, ммоль/л | рСКФ, мл/мин/ 1,73 м2 | ТРФ, % | ТмРФ/ рСКФ, ммоль/л | Нефрокальциноз, степень |
|---|---|---|---|---|---|---|---|---|---|
1 | 42 | <300 | 0,76 | 1,08 | 87 | 71 | 48,8 | 0,53 | 1 |
2§ | 36 | <300 | 7,4 | 1,06 | 81 | 0 | 84,1 | 0,89 | 1 |
3 | 51 | <300 | 2,31 | 1,19 | 67 | 1 | 4,4 | 1,01 | 1 |
4¥ | 35 | 3970 | 8,12 | 1,3 | 77 | 6 | 1,9 | 1,07 | 1 |
5¥ | 28 | 60800 | 5,04 | 1,12 | 82 | 4 | 2 | 0,81 | 1 |
6 | 29 | <300 | 2,4 | 1,38 | 42 | 133 | 93,2 | 1,09 | - |
7 | 36 | <300 | 0,37 | 1,12 | 77 | 86 | 81,2 | 0,86 | - |
8 | 35 | <300 | 3,8 | 0,9 | 75 | 89 | 78,8 | 0,83 | - |
9 | 35 | <300 | 4,1 | 1,41 | 70 | 97 | 89,5 | 1,2 | 1 |
Снижение функций почек до ХБП 2 стадии установлено у 6 (66,7%) женщин - носителей болезни Дента. Медиана рСКФ у матерей - гетерозиготных носителей заболевания была статистически значимо выше по сравнению с их сыновьями с болезнью Дента, обследованными в возрасте 12 (8,1; 15,1) лет: 86,0 (82,0; 94,0) пр. 74,0 (66,5; 82,0) мл/мин/1,73 м2 (p=0,022) (рисунок). Выявлена прямая корреляционная взаимосвязь между рСКФ при последнем обследовании у матерей - носителей CLCN5 и OCRL мутаций и у мальчиков с болезнью Дента: r=0,676 (p=0,025).
Ни у одной из обследованных женщин - носителей болезни Дента не выявлено бессимптомное носительство мутаций в генах CLCN5 и OCRL. Полный фенотип в виде низкомолекулярной протеинурии, гиперкальциурии, нарушения реабсорбции фосфатов с гипофосфатемией, медуллярным нефрокальцинозом и прогрессированием в ХБП 2 стадии выявлен у 2 (22,2%) женщин - двоюродных сестер - носителей болезни Дента 1-го типа.
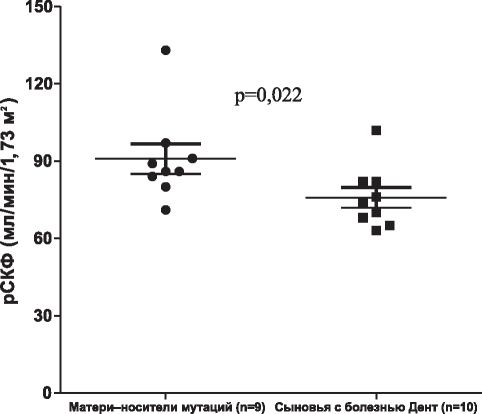
Рисунок. рСКФ у матерей - облигатных носителей мутаций в генах CLCN5 и OCRL и их сыновей с болезнью Дента.
На основании проведенного исследования установлена широкая вариабельность клинических проявлений болезни Дента у всех обследованных матерей мальчиков с данной Х-сцепленной тубулопатией.
Превалирующими клиническими проявлениями болезни Дента, выявленными более чем у 50% мам пациентов, являлись нарушения реабсорбции фосфатов с гипофосфатемией, медуллярный нефрокальциноз и прогрессирование в ХБП 2 стадии. Полученные нами результаты согласуются с данными исследования S.C. Reinhart и соавт. (1995), в котором уролитиаз и снижение рСКФ установлены у 66,7 и 30,8% соответственно женщин - носителей заболевания [13]. В ранее проведенных исследованиях продемонстрировано, что у женщин - гетерозиготных носителей болезни Дента не выявлено нефрокальциноза и/или уро- литиаза, нарушения реабсорбции фосфатов, снижения функционального состояния почек [14].
Нами установлено, что низкомолекулярная протеинурия отмечалась у 22,2% женщин-носителей болезни Дента. По данным ранее проведенных исследований, низкомолекулярная протеинурия выявлялась у 80-100% женщин - носителей болезни Дента [13]. При этом, как и в данном исследовании, протеинурия незначительной степени выраженности обнаружена только у 15,4% мам мальчиков с болезнью Дента [13].
В результате проведенного нами исследования установлено, что гиперкальциурия отмечалась у 22,2% женщин - носителей болезни Дента. Сходные результаты получены в исследовании S.C. Reinhart и соавт. (1995), в котором гиперкальциурия выявлена у 33,3% обследованных женщин-носителей мутаций болезни Дента [13]. По данным исследования O. Wrong (1994), у 50% женщин - носителей болезни Дента определялась гиперкальциурия [26]. Установленные различия в частоте гиперкальциурии среди женщин- носителей могут быть связаны с различным типом мутаций в генах заболевания.
Впервые в литературе нами выявлен полный фенотип болезни Дента 1-го типа у 22,2% обследованных женщин-носителей.
Установленная фенотипическая вариабельность клинических проявлений болезни Дента у всех мам мальчиков с данной Х-сцепленной тубулопатией может быть следствием как аутосомной транслокации - обмена генетической информацией между Х-хромосомой и аутосомой без потери материала, так и неслучайной инактивации Х-хромосомы, при которой соотношение инактивации хромосомы отцовского или материнского Х изменяется в сторону одного из родителей [27].
В результате неслучайной инактивации Х-хромосомы в ранней эмбриональной жизни, у гетерозиготных женщин наблюдается мозаицизм по Х-сцепленной экспрессии гена как от материнской, так и отцовской Х-хромосомы [27]. Поскольку выбор одной или другой Х-хромосомы в начале процесса инактивации обычно является неслучайным [27], значительное отклонение или перекос от ожидаемой средней картины инактивации X-хромосомы (т.е. 50:50) в определенной популяции женщин-носителей предполагает, что X-связанная мутация изменяет жизнеспособность или пролиферацию клеток in vivo [28].
Полученные данные свидетельствуют о необходимости раннего диагностического обследования матерей мальчиков с болезнью Дента с целью определения наличия и выраженности клинических проявлений патологии и своевременной профилактики прогрессирования ХБП.
Таким образом, полученные данные проведенного исследования свидетельствуют о наличии клинических проявлений болезни Дента у всех женщин - носителей мутаций в генах CLCN5 или OCRL, несмотря на Х-сцепленный тип наследования патологии.
Превалирующими клиническими проявлениями болезни Дента у мам пациентов являлись нарушения реабсорбции фосфатов с гипофосфатемией, медуллярный нефрокальциноз и наличие ХББП С2 ст.
Полученные данные свидетельствуют о необходимости диагностического обследования матерей мальчиков с болезнью Дента с целью определения фенотипа и своевременной профилактики прогрессирования ХБП.
1. Dent CE, Friedman M. Hypercalciuric rickets associated with renal tubular damage. Arch Dis Childh 1964;39:240–249
2. Hoopes RR, Raja KM, Koich A et al. Evidence for genetic heterogeneity in Dent's disease. Kidney Int 2004;65:1615–1620
3. Lloyd SE, Gunther W, Pearce SH et al. Characterisation of renal chloride channel, CLCN5, mutations in hypercalciuric nephrolithiasis (kidney stones) disorders. Hum Mol Genet 1997; 6:1233–1239
4. Hoopes RR, Shrimpton AE, Knohl SJ et al. Dent disease with mutations in OCRL1. Am J Hum Genet 2005;76:260–267
5. Anglani F, D'Angelo A, Bertizzolo LM et al. Nephrolithiasis, kidney failure and bone disorders in Dent disease patients with and without CLCN5 mutations. Springerplus 2015;4:492
6. Devuyst O, Thakker RV. Dent’s disease. Orphanet J Rare Dis 2010;14;5:28
7. Zaniew M, Mizerska-Wasiak M, Załuska-Leśniewska I et al. Dent disease in Poland: what we have learned so far? Int Urol Nephrol 2017;49(11):2005–2017
8. Berkel Yv, Ludwig M, Wijk JAE, Bökenkamp A. Proteinuria in Dent disease: a review of the literature. Pediatr Nephrol 2017;32:1851–1859
9. Claverie-Martín F, Ramos-Trujillo E, García-Nieto V. Dent's disease: clinical features and molecular basis. Pediatr Nephrol 2011;26(5):693–704
10. Ludwig M, Utsch B, Balluch B et al. Hypercalciuria in patients with CLCN5 mutations. Pediatr Nephrol 2006;21:1241–1250
11. Bökenkamp A, Böckenhauer D, Cheong HI et al. Dent-2 disease: a mild variant of Lowe syndrome. J Pediatr 2009; 155(1): 94–99
12. Smith AJ. Characterization of Dent's disease mutations of CLC-5 reveals a correlation between functional and cell biological consequences and protein structure. Am J Physiol Renal Physiol 2009;296: 390–397
13. Reinhart SC, Norden AG, Lapsley M. et al. Characterization of carrier females and affected males with X-linked recessive nephrolithiasis. J Am Soc Nephrol 1995;5:1451–1461
14. Scheinman SJ. X-linked hypercalciuric nephrolithiasis: Clinical syndromes and chloride channel mutations. Kidney Int 1998; 53:3–17
15. Norden AG, Scheinman SJ, Deschodt-Lanckman MM et al. Tubular proteinuria defined by a study of Dent's (CLCN5 mutation) and other tubular diseases. Kidney Int 2000;57(1):240–249
16. Edvardsson VO, Goldfarb DS, Lieske JC et al. Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol 2013;28(10):1923–1942
17. Foley KF, Boccuzzi L. Urine Calcium: Laboratory Measurement and Clinical Utility. Lab Medicine 2010;41:683–686
18. Prie D, Ravery V, Boccon-Gibod L, Friedlander G. Frequency of renal phosphate leak among patients with calcium nephrolithiasis. Kidney Int 2001;60:272–276
19. Payne RB. Renal tubular reabsorption of phosphate (TmP/GFR): indications and interpretation. Ann Clin Biochem 1998; 35:201–206
20. Manghat P. Phosphate homeostasis and disorders. Ann Clin Biochem 2014;51:631–656
21. Ronnefarth G, Misselwitz J, members of the Arbeitsgemeinschaft for Padiatrische Nephrologie. Nephrocalcinosis in children: a retrospective survey. Pediatr Nephrol 2000; 14:1016–1021
22. Levey AS, Stevens LA. Estimating GFR using the CKD Epidemiology Collaboration (CKD-EPI) creatinine equation: more accurate GFR estimates, lower CKD prevalence estimates, and better risk predictions. Am J Kidney Dis 2010;55(4):622–627
23. Schwartz GJ, Work DF. Measurement and estimation of GFR in children and adolescents. J Am Soc Nephrol 2009; 4(11): 1832–643
24. National Kidney Foundation Kidney Disease Outcomes Quality Initiatives. K/DOQI Clinical Practice Guidelines for Chronic Kidney Disease Evaluation Classification Stratification. Am J Kidney Dis 2002;39:1–266
25. Sekine T, Nozu K, Iyengar R. et al. OCRL1 Mutations in patients with Dent disease phenotype in Japan. Pediatr Nephrol 2007; 22: 975–980
26. Wrong OM, Norden AG, Feest TG. Dent's disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. Q J Med 1994;87:473–493
27. Lyon MF. Sex chromatin and gene action in the mammalian X chromosome. Am J Hum Genet 1962;14:135–148 28. Lyon MF. Chromosomal and subchromosomal inactivation. Ann Rev Genet 1968;2:31–52
Баширова Зиля Рамилевна, Научно-исследовательский клинический институт педиатрии им. акад. Ю.Е. Вельтищева РНИМУ им. Н.И. Пирогова, отдел наследственных и приобретенных болезней почек
Людвиг Михаэль, доктор медицинских наук, профессор, Молекулярно-биологическая лаборатория, отдел геномики
, 53127, г. Бонн, ул. Sigmund-Freud-Srabe, 25
Приходина Л.С., Папиж С.В., Баширова З.Р., Людвиг М. ЯВЛЯЮТСЯ ЛИ МАМЫ МАЛЬЧИКОВ С БОЛЕЗНЬЮ ДЕНТА БЕССИМПТОМНЫМИ НОСИТЕЛЯМИ Х-СЦЕПЛЕННОЙ ТУБУЛОПАТИИ? Нефрология. 2018;22(2):74-80. https://doi.org/10.24884/1561-6274-2018-22-2-74-80
Prikhodina L.S., Papizh S.V., Bashirova Z.R., Ludwig M. ARE MOTHERS OF BOYS WITH DENT’S DISEASE ASYMPTOMATIC CARRIERS FOR X-LINKED TUBULAR DISORDER? Nephrology (Saint-Petersburg). 2018;22(2):74-80. (In Russ.) https://doi.org/10.24884/1561-6274-2018-22-2-74-80
![]()

197101, Санкт-Петербург, улица Льва Толстого, дом 17, литер А,
редакция журнала «Нефрология»
e-mail: journal@nephrolog.ru



































