


Содержание
Перейти к:
https://doi.org/10.24884/1561-6274-2017-21-6-78-85
Перейти к:
AL-амилоидоз –заболевание, связанное с пролиферацией патологического клона плазматических клеток и экспрессией свободных легких цепей. Формирование специфической амилоидной структуры, состоящей из аберрантных молекул, лежит в основе изменений мультиорганных структурно-функциональных изменений. Типично вовлечение миокарда с развитием рестриктивной кардиомиопатии и прогрессирующей сердечной недостаточностью, а также почек с прогрессирующей протеинурией и дисфункцией. Выявление депозитов амилоида, оценка его состава и выявление моноклональной продукции легкой цепи иммуноглобулина диагностики заболевания. В данной статье приведено описание клинического случая течения, диагностики и успешной терапии AL-амилоидоза с необычным дебютом в виде поражения легких и плевры с дальнейшим вовлечением сердца и почек.
Болдуева С.А., Облавацкий Д.В., Грохотова В.В., Быстрова О.Б., Майер Д.А., Добронравов В.А. КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ СИСТЕМНОГО AL-АМИЛОИДОЗА С НЕОБЫЧНЫМ ДЕБЮТОМ ЗАБОЛЕВАНИЯ. Нефрология. 2017;21(6):78-85. https://doi.org/10.24884/1561-6274-2017-21-6-78-85
Boldueva S.A., Oblavatskii D.V., Grokhotova V.V., Bystrova O.B., Mayer D.A., Dobronravov V.A. A CLINICAL CASE OF SYSTEMIC AL-AMYLOIDOSIS WITH PECULIAR DISEASE PRESENTATION. Nephrology (Saint-Petersburg). 2017;21(6):78-85. (In Russ.) https://doi.org/10.24884/1561-6274-2017-21-6-78-85
Амилоидоз - заболевание, в процессе которого происходит нарушение белкового обмена, приводящее к синтезу и отложению в тканях специфического белка - амилоида. Из многообразия форм системного амилоидоза в соответствии с типом синтезирующегося амилоида наиболее часто диагностируются AL- амилоидоз (ассоциированный с плазмоклеточными дискразиями) и АА-амилоидоз (вторичный амилоидоз при хронических воспалительных заболеваниях; семейный амилоидоз при периодической болезни) [1, 2]. Распространенность АL-амилоидоза, по данным Национального центра медицинской статистики США, составляет 4,5 случая на 100 000, а заболеваемость, примерно, 3200 новых случаев в год [3]. В развивающихся странах смертность от AL-амилоидоза составляет 1 случай на 2000 населения (0,05%) [4]. Точных данных о распространенности заболевания в Российской Федерации нет, а чаще регистрируются случаи вторичного амилоидоза на фоне хрониче-
ских воспалительных заболеваний [5, 6]. В связи с тем, что амилоидоз приводит к мультиорганным поражениям, клиническая картина заболевания бывает весьма разнообразной. Трудности диагностики данного заболевания объясняются многообразием клинических проявлений и симптомов, на первый взгляд, казалось бы, не связанных между собой. Зачастую своевременная постановка диагноза играет решающую роль в прогнозе пациентов. Поэтому представляется целесообразным поделиться полученным клиническим опытом и описать случай системного АL-амилоидоза с необычным дебютом заболевания.
Мужчина, 57 лет, поступил планово 11 ноября 2015 г. в отделение кардиологии клиник СЗГМУ им. И.И. Мечникова из Южного федерального округа РФ с целью обследования и уточнения диагноза выраженной кардиальной дисфункции. При поступлении предъявлял жалобы на общую слабость, быструю утомляемость, снижение аппетита, одышку смешанного характера при небольших нагрузках, отеки нижних конечностей. При активном расспросе по системам органов отмечал малопродуктивный кашель с эпизодами кровохарканья, а также периодическое окрашивание мочи в розовый цвет.
Из анамнеза: в начале 2013 года появились немотивированная слабость, утомляемость, снижение аппетита и массы тела. С декабря 2013 года пациент отметил значительное снижение переносимости нагрузок, периодическое окрашивание мочи в розовый цвет. Весной 2014 года в связи с выявленным двусторонним гидротораксом госпитализирован в торакальное отделение городского стационара по месту жительства. Выполнялись многочисленные плевральные пункции по поводу рецидивирующего гидроторакса. По результатам мультиспиральной компьютерной томографии (МСКТ) органов грудной клетки от марта 2014 г. выявлены признаки хронического эндобронхита, диффузный пневмосклероз без очаговых и инфильтративных изменений легочной ткани, внутригрудной лимфоаденопатии и новообразований. Выполнено множество исследований, исключивших системные заболевания соединительной ткани, артерииты, васкулиты, онкозаболевания, туберкулез. 03.04.2014 г. проведена диагностическая видеоторакоскопия с одномоментной костальной плеврэктомией и ревизией правого легкого, в ткани которого патологии не обнаружено. При гистологическом исследовании костальной плевры - ткань со склерозом, полнокровными сосудами, очаговыми кровоизлияниями, очаговой лимфолейкоцитарной инфильтрацией. Атипичных клеток не выявлено.
С конца апреля 2014 г. пациент отметил резкое ухудшение самочувствия в виде нарастания одышки, появления приступов удушья. 22.04.2014 г. госпитализирован в отделение пульмонологии городского стационара по месту жительства. По результатам рентгенографии органов грудной клетки - абсцедирующая правосторонняя нижнедолевая пневмония. При ангиопульмонографии был подтвержден диагноз инфаркт-пневмонии вследствие перенесенной тромбоэмболии легочной артерии (ТЭЛА) с исходом в абсцесс нижней доли правого легкого. При фибробронхоскопии - признаки хронического катарального эндобронхита, органической патологии не выявлено. По результатам исследования эндобронхиальных смывов - прозрачная бесцветная жидкость с единичными лейкоцитами и лимфоцитами в поле зрения, нормальными клетками цилиндрического и альвеолярного эпителия. В анализах мокроты - признаки гнойного воспаления, альвеолярные макрофаги в умеренном количестве при отрицательной бактериологии. Кислотоустойчивых микроогранизмов не выявлено. В связи с формированием множества абсцессов нижней доли правого легкого 03.06.2014 г. выполнена операция торакотомии, нижней лобэктомии справа. При гистологическом исследовании ткань легкого с диффузным пневмосклерозом, очаговой нейтрофиль- ной и лимфоцитарной реакцией, отложением гемосидерина, множественными кровоизлияниями и микроателектазами. Послеоперационный период без осложнений.
Ввиду наличия симптомов хронической сердечной недостаточности (ХСН), впервые проводится эхокардиография (ЭХО-КГ), по результатам которой камеры сердца не расширены, зон нарушения локальной сократимости, снижения систолической функции, нарушений внутрисердечной гемодинамики, признаков легочной гипертензии не обнаружено. Выявлены умеренная «концентрическая гипертрофия стенок миокарда левого желудочка» (без наличия в анамнезе артериальной гипертензии) и выраженная диастолическая дисфункция миокарда ЛЖ по типу нарушения релаксации. На фоне проводимого консервативного лечения ХСН с положительной динамикой в июле 2014 года пациент выписан на амбулаторное лечение с диагнозом: рестриктивная кардиомиопатия неуточненного генеза. Рекомендована плановая госпитализация в городской ФЦССХ для проведения дополнительного обследования, уточнения диагноза.
В ноябре 2014 года - ухудшение состояния, прогрессирование явлений бивентрикулярной ХСН. В плановом порядке поступает на обследование в ФЦССХ по месту жительства. По результатам лабораторных исследований: в клиническом анализе крови отмечено нарастание СОЭ до 50 мм/ч при нормальном уровне эритроцитов, гемоглобина, тромбоцитов и неизмененной лейкоцитарной формуле. В биохимическом анализе крови выявлены: гипопротеинемия и гипоальбуминемия: общий белок - 49 г/л, альбумин - 28 г/л; нарастание уровней СРБ до 17,3 мг/л, фибриногена до 9 г/л. Впервые отмечено повышение уровней креатинина - 128 мкмоль/л и мочевины - 8,9 ммоль/л, снижение расчетной скорости клубочковой фильтрации (СКФ) до 53 мл/мин. В общем анализе мочи появление протеинурии до 1,0 г/л, нарастание микрогематурии, появление цилиндрурии. Суточная протеинурия - 2,7 г, при полуколичественном исследовании обнаружен белок Бенс-Джонса (один +).
Учитывая клиническую симптоматику преимущественного поражения легких (рецидивирующий гидроторакс, абсцедирующая пневмония в анамнезе) и дисфункции почек, а также ввиду наличия периваскулярного гранулематозного воспаления и наличия единичных гигантских клеток, обнаруженных при пересмотре биопсийного материала ткани плевры, был заподозрен диагноз гранулематоза Вегенера. Последний не подтвердился по результатам негативных исследований антинейтрофильных цитоплазматических антител, включая антитела IgG к эластазе и катепсину.
Принимая во внимание системность поражения с вовлечением почек и обнаружение в анализах мочи белка Бенс-Джонса, обсуждался диагноз миеломной болезни. Отсутствие М-градиента по данным электрофореза сыворотки крови, отсутствие поражения плоских костей скелета, нормальный уровень гемоглобина, отсутствие изменений со стороны лейкоцитарной формулы, а также результаты миелограммы (плазмоциты 7,2%) - предположение не подтвердилось.
21.11.2014 г. проведена магнитно-резонансная томография (МРТ) сердца с контрастированием: выраженная концентрическая гипертрофия миокарда обоих желудочков со снижением КСО ЛЖ до 35 мл, дилатация правого и левого предсердий, диффузное усиление сигнала от миокарда желудочков по типу свечения. Заключение: данные МРТ свидетельствуют о наличии рестриктивной кардиомиопатии. Для исключения амилоидоза 11.2014 г. выполнена биопсия десневого сосочка: слизистая оболочка с отеком и полнокровием. Отложений амилоида не обнаружено.
По результатам обследования высказано представление в пользу наличия у пациента системного неуточненного васкулита, АНЦА-отрицательного, с поражением легких, почек, сердца. Принято решение о проведении терапии глюкокортикоидами (ГКС).
На фоне длительной терапии ГКС в дозе 10 мг/ сут отмечалась некоторая положительная динамика в виде улучшения общего самочувствия, уменьшения слабости, отеков. Состояние пациента оставалось относительно стабильным до сентября 2015 г., однако, диагноз по-прежнему оставался неясным. Для дальнейшего обследования и уточнения диагноза 11 ноября 2015 г. пациент поступает в отделение кардиологии СЗГМУ им. И.И. Мечникова.
Учитывая системность проявлений (сердце, легкие, плевра, почки, гепатоспленомегалия), а также характер поражения сердца (прогрессивное утолщение стенок как ЛЖ, так и ПЖ при отсутствии в анамнезе АГ, увеличение предсердий, рестриктивный тип кардиомиопатии, свечение при МРТ), в первую очередь было решено исключить системный амилоидоз. Менее вероятным представлялся диагноз системного васкулита.
12.11.2015 г. выполнена ЭХО-КГ (рис.1), по данным которой в сравнении с результатами предыдущего исследования от июня 2014 г. - нарастание концентрической гипертрофии стенок ЛЖ (МЖП утолщена до 23 мм, ЗС - 21 мм), гипертрофии стенки ПЖ до 8,3 мм. Выраженная дилатация полостей предсердий. Глобальная сократимость миокарда левого желудочка - ФВ 50% по Симпсону. Рестриктивный тип диастолической дисфункции миокарда. В остальном - без существенной динамики. Также выявлено выраженное усиление ЭХО-сигнала от миокарда «по типу свечения».
Выполнено исследование сыворотки крови на наличие легких цепей иммуноглобулинов, которые позволили выявить наличие лямбда-легких цепей в титре 175 мкг/мл (при норме до 10 мкг/мл). Уровень каппа-легких цепей в пределах нормы.
13.11.2015 г. проведена биопсия миокарда (гистологическое исследование биоптата выполнено в Федеральном центре сердца, крови и эндокринологии им. В.А. Алмазова д-ром мед. наук Г.Б. Митрофановой). По результатам гистологического исследования выявлены умеренная гипертрофия мышечных волокон, фиброз эндокарда, мелкоочаговый субэндокардиальный фиброз, утолщение стенок артерий за счет отложения белковых гомогенных масс. Обнаружено отложение белка с тинкториальными свойствами амилоида в стенках артерий, между волокнами миокарда и в эндокарде: окраска с использованием конго красного с положительным зеленым свечением в поляризованном свете (рис. 2). При иммуногистохимическом исследовании признаков миокардита не выявлено, в зонах отложения амилоида иммуногистохимически была выявлена экспрессия легких цепей лямбда.
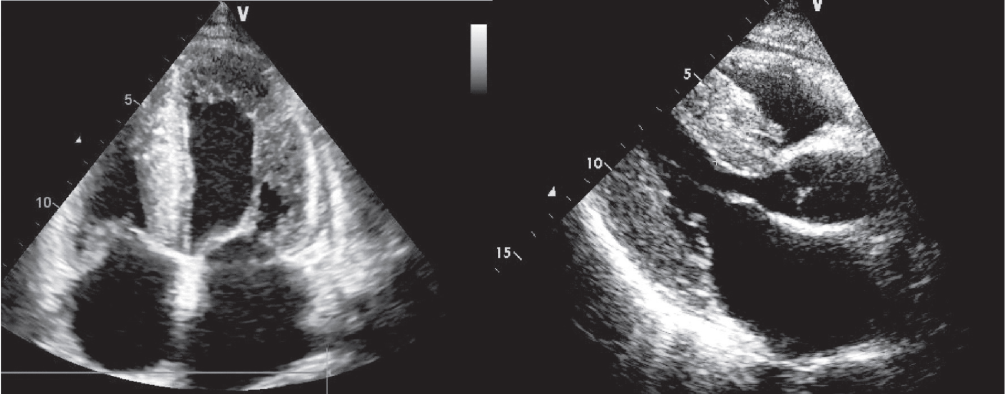
Рис. 1. Эхокардиография (пояснения в тексте).
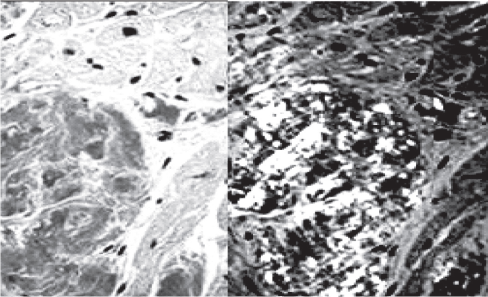
Рис. 2. Отложение амилоида между волокнами миокарда (слева); справа - тот же препарат с положительным желтозеленым свечением в поляризованном свете. Окраска конго красным.
Таким образом, с помощью биопсии миокарда, гистологического и иммуногистохимического исследований подозрение о ПА нашло подтверждение.
Для проведения трепанобиопсии и дальнейшей специфической терапии больной был переведен в центр лечения амилоидоза на базе НИИ нефрологии ПСПбГМУ им. акад. И.П. Павлова.
При поступлении пациента в клинику НИИ нефрологии функция почек была умеренно снижена и соответствовала 3а стадии хронической болезни почек (ХБП) - уровень креатинина 125 мкмоль/л, расчетная СКФ по формуле CKD-EPI составила 55 мл/мин/1,73 м2. Отмечали выраженную проте- инурию (3,5 г/сут), гипоальбуминемию (30,5 г/л), отеки нижних конечностей до уровня нижней трети голеней.
Размеры правой и левой почки по результатам сонографии составили 10,6χ5,1 и 11,1*5,7 см соответственно, с умеренным повышением эхогенности паренхимы. При обследовании обращало на себя внимание повышение уровня тропонина I до 0,203 нг/мл (норма до 0,040 нг/мл), КФК МВ до 8,1 нг/мл (норма 0,6-6,3 нг/мл), NT-proBNP до 8046 пг/мл (норма до 125 пг/мл), что указывало на выраженное повреждение миокарда депозитами амилоида.
По результатам морфологического исследования биоптата почки (морфолог, канд. мед. наук В.Г. Сиповский) из 16 клубочков в срезе 7 были полностью склерозированы, выявлялось незначительное сегментарное утолщение базальной мембраны, расширение мезангиального матрикса, умеренная дистрофия канальцев, очаговый интерстициальный фиброз. Обращала на себя внимание незначительная плазмоцитарная инфильтрация интерстиция, а также накопление в стенках артерий конго-позитивных масс с дихроичным свечением в поляризованном свете, методом иммуногистохимии выявлена экспрессия лямбда свободных легких цепей (СЛЦ) в зонах отложения амилоида. Для дополнительного подтверждения системности патологического процесса была выполнена биопсия подкожного жира, по результатам которой 60% мазка занимали отложения амилоида.
Для уточнения активности заболевания методом иммуноферментного анализа был определен уровень СЛЦ в плазме крови и моче, выявлено повышение лямбда цепей в крови до 80,7 мкг/мл (соотношение лямбда/каппа = 16, норма лямбда СЛЦ 5,71-26,3 мкг/мл) и в моче до 16,6 мкг/мл (соотношение лямбда/каппа = 2, норма лямбда СЛЦ до 12,3 мкг/мл). При иммунофиксации мочи и сыворотки крови моноклональных иммуноглобулинов выявлено не было. В биоптате костного мозга при трепанобиопсии плазмоцитоз составил всего 0,6%. При цитофлюорометрии обнаружены клетки с патологическим иммунофенотипом CD45-/CD138+/CD38+/CD56+/CD117- (0,16% от общего числа ядросодержащих клеток). Уровень бета-2-микроглобулина был повышен, 3,85 мг/л (норма 1,42-3,21 мг/л).
Диагноз системного AL-амилоидоза был установлен на основании следующих критериев: депозитов амилоида в миокарде и почке, экспрессирующих легкую цепь иммуноглобулина лямбда; повышения уровня лямбда СЛЦ моноклонального типа в сыворотке крови, а также обнаруженных в красном костном мозге (ККМ) плазмоцитов с аберрантным иммунофенотипом при низком уровне плазмоцитоза костного мозга и отсутствии других критериев множественной миеломы.
Проведена химиотерапия бортезомибом и дексаметазоном в комплексе с препаратами сопроводительного лечения. Всего на протяжении 2016 года было проведено 6 курсов химиотерапии в стандартных дозировках с учетом площади поверхности тела. Первые два курса сопровождали введение циклофосфамида в дозе 400 мг дважды за курс (всего 1600 мг). Начиная с четвертого курса бортезомиб применяли в редуцированной дозировке, ввиду развития ассоциированной с его приемом выраженной периферической полинейропатии. После проведенного лечения был достигнут полный гематологический ответ в виде нормализации уровня лямбда СЛЦ до 11,98 мкг/ мл (норма 5,71-26,3 мкг/мл). От начала терапии произошло уменьшение ИММ с 259 до 186 г/м2, толщины МЖП (с 26,4 до 26,2 мм) и задней стенки ЛЖ (с 24 до 18 мм), с отчетливым увеличением толерантности к физическим нагрузкам. Положительную динамику продемонстрировали другие исследования в виде снижения СОЭ до 15 мм/ч (норма до 10 мм/ч), тропонина I до 0,064 нг/мл (норма до 0,040 нг/мл), NT-proBNP до 2034 пг/мл (норма до 125 пг/мл), креатинина до 114 мкмоль/л (53-115 мкмоль/л), СПБ составила 0,49 г/сут (норма до 0,15 г/сут), уровень альбумина 41,8 г/л (норма 35-50 г/л), по результатам трепанобиопсии обнаружено не более 1 % зрелых плазматических клеток без признаков атипии.
В описанном клиническом случае от момента появления первых неспецифических симптомов заболевания до постановки правильного диагноза прошло более двух лет. Учитывая тот факт, что своевременная постановка правильного диагноза при ПА является одним из ведущих факторов, влияющих на прогноз таких пациентов, очевидно, что подобные сроки диагностики этого заболевания недопустимы. В определенной степени сложность диагностики первичного амилоидоза состоит в многообразии различных и, на первый взгляд, не связанных между собой симптомов. Процесс клинически дебютировал поражением респираторной системы, которое не является редкостью при ПА. Частота вовлечения в патологический процесс респираторной системы при системных амилоидозах составляет около 50%. Поражения плевры при системном AL-амилоидозе составляет приблизительно 6% и характеризуется формированием рецидивирующего выпотного плеврита, зачастую рефрактерного к терапии [7]. Такую симптоматику наблюдали и в данном случае, предполагая, что гнойные осложнения отчасти носили ятрогенный характер. Последние и отсутствие настороженности, вероятно, повели специалистов «по ложному пути», заставив в первую очередь исключать онкопатологию. В противном случае адекватное исследование амилоида в биопсийном материале плевры и легких позволило бы поставить диагноз ПА еще в 2014 году.
Следует отметить, что биопсия слизистых оболочек десны и ЖКТ, в отличие от биопсии почек или миокарда, не всегда дает положительный результат. Так, по разным данным, положительный результат на обнаружение амилоида при биопсии слизистой оболочки десны составляет от 30 до 40%, слизистых оболочек желудочно-кишечного тракта от 50 до 70%, подкожно-жировой клетчатки передней брюшной стенки около 80%, в то время как положительный результат при биопсии почек или сердца близок к 100% [8]. Диагноз амилоидоза устанавливается на основании результатов гистологического и иммуногистохимического исследования и почти всегда требует подтверждающей диагностики, направленной на определение этиологического варианта болезни. Такая диагностика и была проведена после выявления кардиальных депозитов амилоида и иммуногистохимических указаний на их вероятную связь с плазмоклеточной дискразией. В результате был установлен диагноз AL-амилоидоза.
Лечение AL-амилоидоза основывается на представлении об онкогематологической природе данного заболевания, которое связано с моноклональной плазмоклеточной пролиферацией в ККМ. Вместе с тем, особенностью данного варианта плазмоклеточной дискразии является то, что клинико-морфологические проявления AL- амилоидоза не связаны с массой опухолевого клона, но определяются исключительно накоплением в тканях и гиперпродукцией моноклональными клетками аберрантных легких цепей иммуноглобулинов (в обсуждаемом случае - лямбда). Повышение пула этих молекул в плазме крови приводит к их отложению в тканях и формированию бета-складчатой структуры AL-амилоида [9]. Прогноз и эффективность лечения определяются накопившимся объемом тканевых депозитов амилоида и степенью нарушения функций жизненно важных органов [10], поскольку быстрая редукция относительно небольшого клона аберрантных плазмоцитов не является сложной задачей при возможностях современной химиотерапии. Так, и в данном случае вовлеченность в патологический процесс сразу нескольких органов (сердце, почки, легкие, подкожно-жировая клетчатка) свидетельствовала об интенсивной экспрессии патологических легких цепей опухолевыми клетками в костном мозге. Однако содержание плазмоцитов по результатам трепанобиопсии в костном мозге было нормальным. Доказательствами наличия опухолевого клона были выявленный плазмоцитоз 7,2% по результатам ранее выполненной миелограммы и данные проточной цитометрии. Это несоответствие тяжести мультиорганного поражения и активности процесса количеству заинтересованных в нем клеток может объясняться как определенной «гнездностью» расположения очагов плазмоцитарной инфильтрации, так и их внемозговой локализацией. Последняя, вероятно, является особенностью данного случая и была представлена плазмоцитарной инфильтрацией легких, плевры и интерстиция почек по результатам гистологических исследований, проведенных в разное время.
В среднем медиана выживаемости при AL- амилоидозе от постановки диагноза составляет 3 года, этот срок уменьшается до 1 года при клинически значимом вовлечении миокарда [1, 11]. Доминирующие клинические проявления со стороны сердца в виде развития тяжелой сердечной недостаточности за счет резкого увеличения толщины миокарда желудочков и рестриктивных нарушений, а также признаки выраженной реналь- ной дисфункции делали прогноз пациента крайне серьезным.
Терапия данного заболевания направлена на торможение формирования амилоида в тканях за счет воздействия на причину - клон плазматических клеток, ответственный за экспрессию свободных легких цепей. Это достигается применением различных вариантов химиотерапии, включающей протеосомные ингибиторы, глюко- кортикоиды, алкилирующие агенты.
Базовыми препаратами для лечения AL- амилоидоза на сегодняшний день являются средства, блокирующие активность 26S-протеосомы [12]. Этот крупный ферментативный комплекс отвечает за биодеградацию до аминокислот поврежденных белков цитоплазмы, торможение его работы приводит к их накоплению и запуску механизмов апоптоза. В результате происходят нормализация уровня свободных легких цепей иммуноглобулинов в сыворотке крови и прекращение образования амилоида в тканях. Совместно с ингибитором протеосом (бортезомибом) назначают глюкокортикоиды, обладающие неспецифическим иммуносупрессивным свойством, а также циклофосфамид - алкилирующий агент, вызывающий повреждение ДНК и гибель активно делящихся клеток. В заключение следует еще раз остановиться на принципиальной важности своевременной диагностики AL-амилоидоза, онкогематологического заболевания, медиана выживаемости при котором даже с использованием современных методов лечения остается неудовлетворительно низкой. При этом очевидно, что раннее начало терапии приводит к торможению образования амилоида в тканях, а значит и к улучшению прогноза у таких пациентов [13]. При этом гематологическая ремиссия в случаях поздней диагностики заболевания не всегда приводит к улучшению функции пораженных органов ввиду устойчивости AL-амилоида к действию внеклеточных ферментов, а также за счет его непосредственного токсического действия на окружающие клетки [14,15].
В данном случае индукционная терапия дексаметазоном, бортезомибом и циклофосфамидом с продолжением бортезомибом и дексаметазоном привела к ожидаемому результату - достижению полного ответа в виде нормализации концентрации моноклональной легкой цепи в циркуляции. На эффект терапии не повлияла необходимость некоторой редукции доз бортезомиба из-за развития типичного осложнения - полинейропатии. Отчетливым был и достигнутый клинический ответ в виде существенного снижения проявлений сердечной недостаточности на фоне редукции массы миокарда, а также выраженное снижение протеинурии.
Поражение почек при AL-амилоидозе складывается из нескольких компонентов: во-первых, это непосредственное отложение амилоида в структурах клубочка и тубулоинтерстиция с закономерным нарушением их архитектоники и снижением функции [16]. Второй механизм заключается в развитии MIDD (monoclonal immunoglobulin deposition disease) - заболевания, связанного с депозицией легких цепей иммуноглобулинов в структурах гломерулярной и канальцевой базальной мембраны, мезангия, в цитоплазме подоцитов с формированием кристаллических включений, повреждающих цитоскелет, что способствует развитию протеинурии и потенцирует течение гломерулосклероза [17, 18]. Протеинурия в данном клиническом случае носила явный гломерулярный характер. Гистологически в почке амилоид и экспрессия легкой цепи были выявлены только в стенках артерий, что не могло быть причиной протеинурии субнефротического уровня. В отсутствие существенной депозиции амилоида в матриксе клубочка мы предполагаем, что протеинурия была обусловлена интраподоцитарным накоплением легких цепей (которые недоступны выявлению при иммуногистохимическом исследовании) с нарушением цитоскелета подоцито, как особого варианта MIDD. Тканевой клиренс элиминация депозитов AL-амилоида с участием внеклеточных протеаз (катепсины B и L) растягивается на достаточно длительный срок и зависит от степени снижения СЛЦ в циркуляции [19, 20]. Таким образом, становится очевидным, что быстрый почечный ответ на фоне проводимой терапии - выраженное снижение протеинурии - явилось следствием снижения пула свободных легких цепей в циркуляции и снижения аккумуляции их в подоцитах.
Приведенный случай, являясь типичным в плане запоздалого выявления AL-амилоидоза из-за недостаточной настороженности в отношении этой болезни, описывает стандартные подходы к адекватной диагностике и возможности этиотропной терапии в коррекции дисфункции сердца и почек.
1. Kumar SK, Gertz MA, Lacy MQ et al. Recent improvements in survival in primary systemic amyloidosis and the importance of an early mortality risk score. Mayo Clinic Proceedings 2011; 86: 12–18
2. Cacoub P, Axler O, De Zuttere D et al. Amyloidosis and cardiac involvement. Ann Med Int 2000; 151: 611-617
3. Шилова ЕМ. Нефрология. ГЭОТАР-Медиа, М., 2010; 694-696 [Shilova EM. Nefrologiya. GEOTAR-Media, M., 2010; 694-696]
4. Батюшин ММ, Повилайтите ПЕ. Клиническая нефрология. Руководство. ЗАОр НПП Джангар, Элиста, 2009; 656 [Batsushin, Poviliteit PE. Clinical nephrology. ZAOr NPP Jangar, Elista, 2009; 656]
5. Волкова ЕН, Посненкова ОМ, Попова ЮВ, Киселев АP. Бюллетень медицинских интернет-конференций. 2014: 4: 1038 [Volkova EN, Posnenova OM, Popova YuV, Kiselyov AP. Bulleten meditcinskikh internet conferentcii. 2014: 4: 1038]
6. Орлова НВ, Гундорова ЛВ, Кривоносов ВВ. Клинический случай: ПА с преимущественным поражением сердца. Лечебное дело 2013; 3: 85-86 [OrlovaNV, GundorovaLV, KrivonosovVV. Clinitcheskiy slutchai: PA s preimuschestvennym porazheniem serdtca. Letschebnoe delo 2013; 3: 85-86]
7. Berk JL, Keane J, Seldin DC et al. Persistent pleural effusions in primary systemic amyloidosis. Chest 2003; 124: 969-977
8. Козловская ЛВ, Рамеев ВВ. Проект клинических рекомендаций по диагностике и лечению системного амилоидоза. Научное общество нефрологов России, М., 2014:13-15 [KozlovskayaLV, Rameev VV. Proekt klinitscheskih recomendatciy po diagnostike I letscheniyu sistemnogo amiloidoza. Nautschnoe obschestvo nefrologov Rossii. M., 2014: 13-15]
9. Bhat A, Selmi C, Naguwa SM et al. Currents concepts on the immunopathology of amyloidosis. Clin Rev Allergy Immunol 2010; 38(2-3): 97-106
10. Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med1997; 337: 898–909
11. Dispenzieri A, Gertz MA, Kyle RA et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol 2004 Sep 15; 22(18): 3751-3757
12. Bonvini P, Zorzi E, Basso G et al. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma. Leukemia 21 (4): 838–842
13. Grogan M, Dispenzieri A, Gertz MA. Light-chain cardiac amyloidosis: strategies to promote early diagnosis and cardiac response. Heart 2017 Jul;103(14):1065-1072
14. McWilliams-Koeppen HP, Foster JS, Hackenbrack N et al. Light Chain Amyloid Fibrils Cause Metabolic Dysfunction in Human Cardiomyocytes. PLoS One 2015 Sep 22; 10(9):e0137716
15. Guan J, Mishra S, Qiu Y et al. Lysosomal dysfunction and impaired autophagy underlie the pathogenesis of amyloidogenic light chain-mediated cardiotoxicity. EMBO Mol Med 2014 Nov; 6(11):1493-1507
16. Desport E, Bridoux F, Sirac C et al. Al amyloidosis. Orphanet J Rare Dis 2012 Aug 21;7:54. doi: 10.1186/1750-1172-7-54
17. Cohen C, Royer B, Javaugue V et al. Bortezomib produces high hematological response rates with prolonged renal survival in monoclonal immunoglobulin deposition disease. Kidney Int 2015 Nov;88(5):1135-1143. doi: 10.1038/ki.2015.201
18. Akilesh S, Alem A, Nicosia RF. Combined crystalline podocytopathy and tubulopathy associated with multiple myeloma. Hum Pathol 2014 Apr;45(4):875-879. doi: 10.1016/j.humpath.2013.10.007
19. Ingrid I. van Gameren, Martin H. van Rijswijk, Johan Bijzet et al. Histological regression of amyloid in AL amyloidosis is exclusively seen after normalization of serum free light chain. Haematologica 2009 Aug; 94(8): 1094–1100. doi: 10.3324/haematol.2008.004119
20. Bohne S, Sletten K, Menard R et al. Cleavage of AL amyloid proteins and AL amyloid deposits by cathepsins B, K, and L. J Pathol 2004 May;203(1):528-537
Болдуева Светлана Афанасьевна - доктор медицинских наук, профессор, Кафедра факультетской терапии.
195067, Санкт-Петербург, Пискаревский пр., д. 47, 8(812)543-15-71
Облавацкий Дмитрий Вячеславович - врач-кардиолог, Отделение кардиологии для лечения больных с инфарктом миокарда.
195067, Санкт-Петербург, Пискаревский пр., д. 47, 8(812)543-15-71
Грохотова Вера Владимировна - кардиолог-аритмолог, Отделение хирургического лечения сложных нарушений сердечного ритма.
195067, Санкт-Петербург, Пискаревский пр., д. 47, 8(812)543-16-16
Быстрова Ольга Борисовна - врач-нефролог.
8(952)202-47-24
Майер Дмитрий Андреевич - клинический ординатор.
8(981)710-01-98
Добронравов Владимир Александрович - доктор медицинских наук, проф., заместитель директора НИИ нефрологии по научной работе.
+7(812)338-69-16
Болдуева С.А., Облавацкий Д.В., Грохотова В.В., Быстрова О.Б., Майер Д.А., Добронравов В.А. КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ СИСТЕМНОГО AL-АМИЛОИДОЗА С НЕОБЫЧНЫМ ДЕБЮТОМ ЗАБОЛЕВАНИЯ. Нефрология. 2017;21(6):78-85. https://doi.org/10.24884/1561-6274-2017-21-6-78-85
Boldueva S.A., Oblavatskii D.V., Grokhotova V.V., Bystrova O.B., Mayer D.A., Dobronravov V.A. A CLINICAL CASE OF SYSTEMIC AL-AMYLOIDOSIS WITH PECULIAR DISEASE PRESENTATION. Nephrology (Saint-Petersburg). 2017;21(6):78-85. (In Russ.) https://doi.org/10.24884/1561-6274-2017-21-6-78-85
![]()

197101, Санкт-Петербург, улица Льва Толстого, дом 17, литер А,
редакция журнала «Нефрология»
e-mail: journal@nephrolog.ru



































