


Содержание
Перейти к:
https://doi.org/10.24884/1561-6274-2018-22-2-30-38
Перейти к:
Рост интереса нефрологов к проблеме поражения почек при преэклампсии (ПЭ) обусловлен её высокой распространенностью (2–14% от всех беременностей) и непредсказуемым исходом. Патогенез поражения почек при ПЭ мало изучен, однако, установлена роль дисбаланса про- и антиангиогенных факторов – VEGF и sFlt-1 – как одного из важнейших патогенетических механизмов, лежащих в основе развития ПЭ. В статье приводятся современные данные о патогенезе и почечных проявлениях ПЭ на разных сроках её развития в сопоставлении с маркерами плацентарного ангиогенеза. Обсуждается роль дисбаланса системы sFlt-1/VEGF в формировании протеинурии, артериальной гипертензии и ренальной дисфункции при ПЭ, а также «ранней» ПЭ как фактора риска развития хронической болезни почек (ХБП).
Меркушева Л.И., Козловская Н.Л. ПОРАЖЕНИЕ ПОЧЕК ПРИ ПРЕЭКЛАМПСИИ: ВЗГЛЯД НЕФРОЛОГА (ОБЗОР ЛИТЕРАТУРЫ). Нефрология. 2018;22(2):30-38. https://doi.org/10.24884/1561-6274-2018-22-2-30-38
Merkusheva L.I., Kozlovskaya N.L. RENAL INJURY IN PREECLAMPSIA: THE VIEW OF NEPHROLOGIST (LITERATURE REVIEW). Nephrology (Saint-Petersburg). 2018;22(2):30-38. (In Russ.) https://doi.org/10.24884/1561-6274-2018-22-2-30-38
Преэклампсия (ПЭ), осложняющая 5-10% всех беременностей, до настоящего времени остается основной причиной материнской и перинатальной смертности [1]. Ежегодно в мире регистрируются более 8 млн случаев ПЭ, уносящих жизни 60 тысяч молодых женщин. Столь высокая частота ПЭ дает основания считать ее самой частой причиной гломерулярного поражения почек [2, 3]. Патогенез ПЭ сложен и не до конца расшифрован. Установлено, что в основе развития ПЭ на первом этапе лежит нарушение плацентации вследствие дефекта ремоделирования сосудов миометрия, что приводит к неполноценной инвазии трофобласта на ранних сроках беременности. В дальнейшем поврежденная ишемизированная плацента в избыточном количестве начинает секретировать мощный антиангиогенный фактор - растворимый рецептор к сосудистому эндотелиальному фактору роста (VEGF), идентифицированный как растворимая fms-подобная тирозинкиназа-1 (sFlt-1). Этот фактор ингибирует как VEGF, так и плацентарный фактор роста (PlGF), обеспечивающие нормальное развитие и функцию плаценты, и, циркулируя в кровотоке матери, может вносить свой вклад в развитие системной эндотелиальной дисфункции, лежащей в основе всех клинических проявлений ПЭ [4, 5].
Преэклампсия, наиболее тяжелое гипертензионное осложнение беременности, характеризуется развитием de novo после 20 нед гестации артериальной гипертензии (АГ) и протеинурии. Кроме них, проявлениями ПЭ служат нарушение функции почек и отёки. Поскольку отеки возникают у 60% женщин с физиологически протекающей беременностью, в настоящее время изолированные отёки перестали рассматривать как признак ПЭ. Однако и сегодня ПЭ нередко манифестирует внезапной резкой прибавкой беременной массы тела с отеками кистей рук, стоп и лица [6].
В 2000 г. рабочая группа Национальной программы изучения артериальной гипертензии (National High Blood Pressure Education Program, NHBPEP) подтвердила, что критериями АГ беременных считается повышение АД свыше 140/90 мм рт. ст., измеренное два раза и более в течение 4-6 ч, особенно у женщин после 20 нед гестации с предшествующим нормальным АД. Было также высказано мнение, что повышение САД на 30, а ДАД на 15 мм рт. ст. от исходного даже в случае АД ниже 140/90 мм рт. ст. обязывает наблюдать женщину для исключения развития преэклампсии. Механизмы развития АГ при ПЭ многообразны и включают в себя повышенные периферическое сосудистое сопротивление как результат генерализованной вазоконстрикции, активность симпатической нервной системы, сердечный выброс, избыточный ответ на различные прессорные стимулы, в том числе ангиотензин II, катехоламины, эндотелин [6].
Протеинурия (ПУ) может предшествовать АГ, но обычно развивается одновременно или следом за ней. Важнейшим диагностическим критерием ПЭ считается уровень ПУ, превышающий 0,3 г/ сут. Следует отметить, что в последние годы отношение к ПУ, как обязательному критерию ПЭ, изменилось: диагноз ПЭ может быть установлен в отсутствие мочевого синдрома пациенткам с АГ, развившейся во второй половине беременности, при наличии системных проявлений, тромбоцитопении или HELLP-синдрома, в связи с чем предлагалось совсем исключить ПУ из числа клинических проявлений ПЭ [7]. Тем не менее, с точки зрения нефролога, ПУ остается не только важным проявлением ПЭ, но и имеет в ряде случаев прогностическое значение. Протеинурия отличается быстрым (иногда почасовым) нарастанием. Однако гипопротеинемия (поскольку у беременных концентрация альбумина в крови обычно снижена за счет разведения и составляет не более 30 г/л, о гипопротеинемии говорят при уровне альбумина ниже 25 г/л) и нефротические отеки могут отсутствовать в первые дни развития ПЭ [8]. Преэклампсия считается ведущей причиной развития нефротического синдрома (НС) во время беременности [6], хотя этот факт практически неизвестен нефрологам. Однако еще в 1977 г. К.А. Fisher и соавт., выполнявшие после родоразрешения биопсию почки пациенткам с ПЭ в связи с развитием НС, обнаружили лишь её гистологические признаки в 67% биоптатов [9]. В целом НС служит довольно редким проявлением ПЭ (не более 0,0025%) [10], в связи с чем, по-видимому, до настоящего времени условия его развития не изучены. В нашем исследовании, посвященном сравнению клинических проявлений и ближайших почечных исходов нефропатии у пациенток, не имевших почечной патологии до беременности, ПЭ до 34 нед гестации (1-я группа, «ранняя») уровень протеинурии в среднем был почти в два раза выше, чем при ПЭ после 35 нед (2-я группа, «поздняя»). НС развился у 12 из 28 пациенток 1-й группы и лишь у одной из 15 - 2-й группы (42,7 vs 6,7% соответственно) [11, 12]. Эти результаты позволяют предполагать, что «раннее» развитие ПЭ может быть фактором риска идиопатического НС, обусловленного непосредственно ПЭ. После завершения беременности ПУ обычно исчезает в течение 3-8 нед, однако в ряде случаев может персистировать несколько месяцев. Причины и механизм преэкламптической протеинурии будут изложены ниже.
Еще одним «почечным» проявлением ПЭ служит снижение скорости клубочковой фильтрации (СКФ), на которое, однако, редко обращают внимание не только акушеры, но и нефрологи. При физиологической беременности СКФ увеличивается на 40-60% уже в течение I триместра, достигая 140-170 мл/мин [13], в результате чего в сыворотке крови снижается концентрация креатинина (СКр), мочевой кислоты, мочевины. При развитии ПЭ СКФ снижается на 30-40%, а иногда и более по сравнению с нормальной беременностью, однако уровень сывороточного креатинина (СКр) практически всегда соответствует референсным значениям небеременных женщин и редко превышает 90-100 мкмоль/л [14], что, в свою очередь, приводит к недооценке состояния пациентки, особенно при небольшой ПУ и отсутствии тяжелой АГ. В связи с этим «золотым стандартом» оценки функции почек во время беременности считается определение СКФ методом Реберга-Тареева. Расчетные формулы неприемлемы для применения у беременных, так как СКФ по формуле MDRD существенно занижает её значения, а формула Кокрофта-Голта, напротив, завышает их [15, 16]. В уже упомянутом исследовании, предпринятом нами, СКФ, определённая методом Реберга-Тареева, составила у здоровых беременных в среднем 142,5 мл/мин, при ранней ПЭ была вдвое ниже - 70 мл/мин, при поздней - 95 мл/мин, причем различия оказались статистически достоверными не только между группой контроля и обеими группами с ПЭ, но и при сравнении последних между собой. Важной отличительной особенностью «ранней» ПЭ оказалась высокая частота ренальной дисфункции, наблюдавшейся в этой подгруппе больных почти в 4 раза чаще, чем при «поздней» ПЭ: 78,6 vs 20% (р<0,05). При этом у трети пациенток с «ранней» ПЭ зафиксировано изолированное снижение СКФ при нормальном уровне СКр [11, 12]. В редких случаях ПЭ снижение СКФ прогрессирует до острого повреждения почек (ОПП).
Другим характерным признаком нарушения функции при ПЭ является прогрессирующее повышение сывороточного уровня мочевой кислоты. Выраженность гиперурикемии не соответствует уровню СКр, но коррелирует с величиной ПУ, тяжестью ПЭ и перинатальной смертностью. Гиперурикемия часто предшествует протеинурии и снижению СКФ. Повышение уровня мочевой кислоты при ПЭ обусловлено не только ухудшением почечной перфузии и ишемией почек, как полагали ранее, но и снижением почечного клиренса [6, 17]. В последние годы появились предположения о том, что у женщин с ПЭ гиперурикемия может вносить непосредственный вклад в развитие сосудистого повреждения и АГ [6].
Морфологической основой ПЭ является гломерулярный капиллярный эндотелиоз. Его характерными особенностями служат отек эндотелиальных клеток клубочков, утрата ими фенестр и отслойка от базальной мембраны, что приводит к сужению просвета капилляров, иногда вплоть до полной окклюзии. Эти изменения позволяют рассматривать поражение почек при ПЭ как особый тип тромботической микроангиопатии (ТМА), несмотря на редкость тромбозов капиллярных петель клубочков при световой микроскопии [6, 18, 19]. При тяжелой или затянувшейся ПЭ, тем не менее, тромбы обнаруживают не только в гломерулярных капиллярах, но и в мелких внегломерулярных сосудах. Наиболее частой находкой при иммунофлюоресцентном исследовании являются депозиты фибрина в капиллярах клубочков, которые чаще всего выявляются при ранней или тяжелой ПЭ [18]. Следует отметить, что в последние годы получены данные о том, что умеренно выраженный эндотелиоз, считающийся ранее патогномоничным признаком ПЭ, выявляется у 1/3 пациенток с гестационной АГ даже в отсутствие признаков ПЭ. Более того, минимальные гистологические признаки эндотелиоза в виде очагового отека эндотелиальных клеток встречаются даже у здоровых беременных. Это позволяет предполагать, что изменения эндотелиальных клеток клубочка, выраженные минимально, могут быть характерны для беременности per se [18, 20].
У больных с ранними и тяжелыми формами ПЭ при биопсии почки в послеродовом периоде в 3571% случаев выявляется фокально-сегментарный гломерулосклероз (ФСГС), носящий, как правило, вторичный характер [21, 22]. Среди причин развития ФСГС в этих случаях обсуждают гломерулярный эндотелиоз, внутриклубочковую гипертензию и гиперкоагуляцию. Совсем недавно было высказано предположение о том, что вторичный ФСГС может быть следствием подоцитопении, обнаруженной у женщин с ПЭ [23]. Течение вторичного ФСГС в исходе ПЭ более благоприятное, чем течение первичного ФСГС. У 20-30% пациенток с выявленным после перенесенной ПЭ ФСГС персистирует АГ, тогда как ПУ отсутствует или выражена минимально. При повторных нефробиопсиях у этих женщин гистологические проявления ФСГС сохраняются даже в отсутствие прогрессирования клинических признаков, хотя явления эндотелиоза постепенно исчезают.
Другой морфологической находкой, обнаруживаемой при тяжелом течении нефропатии у беременных, являются фибриноидный некроз и склероз междольковых артерий почек [Aber G., 1978; ВиМеп R. et al., 1979]. Эти изменения являются результатом прямого повреждающего действия фульминантного развития тяжелой или злокачественной гипертензии в момент ПЭ. В отдаленном послеродовом периоде у 75% женщин со склерозом междольковых артерий сохраняется устойчивая АГ, нередко с признаками озлокачествления.
Очевидно, что именно ФСГС и склероз внутрипочечных артерий лежат в основе «остаточных изменений» после перенесенной ПЭ, протекающих обычно под маской «гипертонической болезни».
Сегодня уже не вызывает сомнений тот факт, что в основе «материнского синдрома» при ПЭ лежит системная эндотелиальная дисфункция, являющаяся результатом дисбаланса факторов ангиогенеза - дефицитом проангиогенных VEGF и PlGF при избытке растворимых рецепторов к VEGF - VEGFR-1, идентифицированных как растворимая fms-подобная тирозинкиназа-1 (sFlt-1) .
VEGF - сигнальный белок, вырабатываемый клетками для стимуляции васкулогенеза (образование эмбриональной сосудистой системы) и ангиогенеза (рост новых сосудов в уже существующей сосудистой системе). Наиболее важную роль в организме человека играет белок семейства VEGF, называемый VEGF-A. В данное семейство также входят плацентарный фактор роста - PlGF и белки VEGF-B (эмбриональный ангиогенез тканей миокарда), VEGF-C (ангиогенез лимфатических сосудов), VEGF-D (развитие лимфатических сосудов в лёгких). Все члены семейства белков VEGF функционируют, связываясь с двумя близкими по строению мембранными тирозинкиназными рецепторами: рецептором-1 VEGF (VEGFR-1 или Flt-1) и рецептором-2 VEGF (VEGFR-2 или Flk-1) и активируя их. Эти рецепторы экспрессируются эндотелиальными клетками [24]. Белок VEGF-A связывается с рецепторами VEGFR-1 и VEGFR-2, при этом рецептор VEGFR-2 выступает как посредник почти во всех известных реакциях клетки на VEGF. VEGFR-1 также может выступать как «пустой» рецептор, изолируя белок VEGF от рецептора VEGFR-2 (что представляется особенно важным при васкулогенезе зародыша).
Экспрессию VEGF стимулируют множество проангиогенных факторов, включая эпидермальный ростовой фактор, основной фактор роста фибробластов, тромбоцитарный фактор роста и интерлейкин-1. Кроме того, уровни VEGF непосредственно регулируются такими факторами гомеостаза, как рН, давление и концентрация кислорода. Общее влияние перечисленных факторов заключается в опосредованной через VEGF стимуляции важных для ангиогенеза веществ, включая антиапоптотические белки, молекулы клеточной адгезии и металлопротеиназы. Однако основным стимулом экспрессии и/или продукции VEGF является гипоксия [25]. В физиологических условиях основными функциями VEGF-А являются: стимуляция пролиферации эндотелиальных клеток и их дифференциации; усиление сосудистой проницаемости; опосредованная эндотелий-зависимая вазодилатация; поддержание жизнеспособности эндотелия путем предотвращения апоптоза эндотелиальных клеток; участие в ремоделировании экстрацеллюлярного матрикса путем индукции экспрессии активатора плазминогена и ингибитора активатора плазмино- гена (PAI); усиление экспрессии молекул адгезии на поверхности эндотелиальных клеток [26].
В 2003 г. S.E. Maynard и соавт. установили наличие дефицита VEGF у пациенток с ПЭ. Оказалось, что в этом случае ингибиция VEGF была вызвана растворимыми рецепторами к VEGF - VEGFR-1 или растворимой fms-подобной тирозинкиназой-1 (sFlt-1), которую синтезирует ишемизированная плацента [5]. Установлено, что избыточный синтез sFlt-1 начинается за 5-6 нед до клинической манифестации ПЭ. Этот фактор ингибирует как VEGF, так и PlGF, обеспечивающий васкулогенез, и, циркулируя в кровотоке матери, может вносить свой вклад в развитие системной эндотелиальной дисфункции, лежащей в основе всех клинических проявлений ПЭ [4, 5]. Роль sFlt-1 была подтверждена и в эксперименте. По данным, полученным на экспериментальных моделях беременных и небеременных крыс, у которых путем введения в вену sFlt-1 вызывали ПЭ, оказалось, что повышенная концентрация в кровотоке sFlt-1 в обеих группах животных индуцировала АГ и протеинурию, что напоминало «человеческую» преэклампсию. Гистологические исследования ткани почек этих животных указывали на наличие гломерулярного эндотелиоза, характерного для ПЭ [5]. При введении рекомбинантного VEGF-121 крысе с клинической картиной ПЭ проявления последней быстро регрессировали, демонстрируя дозозависимый эффект [27]. Эти данные подтверждают результаты совместных исследований корейских и американских ученых, продемонстрировавших в 2001 г. регресс индуцированной почечной ТМА у крыс после введения рекомбинантного VEGF-121. Выводы были подтверждены морфологически [28]. Таким образом, блокада VEGF является одним из важнейших патогенетических механизмов гломерулярного повреждения, лежащего в основе «нефрологической» составляющей ПЭ [29, 30].
В почках VEGF экспрессируется подоцитами, а рецепторы к нему - эндотелиальными клетками клубочков, что обусловливает важнейшее локально-почечное значение VEGF. Его паракринная функция в отношении эндотелиальных клеток обеспечивает регуляцию клубочковой проницаемости, образование и поддержание фенестрации эндотелия капилляров клубочков [31]. Оказалось также, что подоцитарный VEGF обладает не только паракринной, но и аутокринной функцией - в отношении самих подоцитов, обеспечивающей поддержание их цитоскелета [32].
Предпосылками для изучения «почечных» эффектов факторов ангиогенеза при ПЭ явилось сходство клинических проявлений последней и побочных эффектов анти- VEGF-терапии при лечении злокачественных опухолей, которая, как оказалось, может индуцировать развитие ренальной тромботической микроангиопатии (ТМА) [18, 32, 33]. Первые описания ТМА, как побочного эффекта анти- VEGF-терапии, появились в начале нашего века. Так, у ряда пациентов с раком почки применение бевацизумаба - моноклональных антител к VEGF - приводило к АГ и протеинурии [34]. Позже V. Eremina и соавт. представили шесть клинических наблюдений нефропатии у пациентов со злокачественными новообразованиями разных локализаций, получавших терапию бевацизумабом. Авторы оценивали функцию почек, уровень суточной ПУ до начала и в ходе лечения препаратом. У всех пациентов, не имевших ранее признаков поражения почек и АГ, констатировано нарушение функции почек, нарастающая ПУ и развитие АГ в ближайшие месяцы от начала терапии, что явилось показанием к выполнению биопсии почки. Во всех шести нефробиоптатах выявлена картина ТМА, сочетающаяся с распластыванием малых отростков подоцитов, более выраженным у больных с массивной ПУ. После отмены бевацизумаба ПУ исчезла, функция почек нормализовалась у всех пациентов [29]. Авторы высказали предположение, что снижение уровня VEGF в почке в результате блокады его антителами могло привести к локальной почечной дисфункции эндотелия (у всех больных имелись лишь почечные проявления ТМА и отсутствовали её системные проявления) вследствие нарушения взаимодействия VEGF со своими рецепторами, экспрессируемыми эндотелиальными клетками клубочков. Другими представителями анти-VEGF-препаратов стали ингибиторы тирозинкиназы, первыми из которых были сунитиниб и сорафениб, блокирующие рецепторы к VEGF и также приводящие к развитию почечной ТМА [30, 35]. Сходство клинических проявлений побочных эффектов анти-VEGF-терапии и ПЭ дало основание назвать ренальные последствия терапии ингибиторами ангиогенеза «преэклампсия-подобным синдромом» [36] и стало новым импульсом к изучению механизмов развития основных «почечных» симптомов ПЭ.
Сегодня получены убедительные доказательства того, что при ПЭ дефицит гломерулярного VEGF, обусловленный его блокадой вследствие связывания sFlt-1, в избытке циркулирующем в кровотоке беременной, играет ключевую роль не только в генезе ренальной дисфункции и АГ, но также ПУ. В предпринятом нами исследовании по сравнению с ранней и поздней ПЭ мы также изучали содержание sFlt-1 и VEGF в этих группах беременных. Оказалось, что у женщин с ранней ПЭ, имевших более тяжелую АГ, более низкую СКФ и более выраженную протеинурию, уровень sFlt-1 был не только выше, чем у здоровых беременных, но троекратно превышал таковой у пациенток с поздней ПЭ, причем СКФ обратно коррелировала с содержанием sFlt-1, что позволяет обсуждать связь этого маркера с состоянием функции почек [11, 12].
Основной причиной уменьшения СКФ, как было установлено, является утрата фенестр клетками гломерулярного эндотелия. Поскольку для образования и поддержания фенестрации необходим VEGF, очевидно, что результатом дефицита последнего станет нарушение структуры и функции эндотелия (гломерулярный эндотелиоз) с последующим снижением СКФ [19, 32]. Индуцированное блокадой VEGF эндотелиальное повреждение может также быть причиной артериальной гипертензии. Связь последней с дефицитом VEGF была продемонстрирована в исследовании E.Robinson и соавт., установившими, что ингибиция VEGF противоопухолевым препаратом седиранибом - сильнодействующим ингибитором VEGFR-2 - в течение трех дней индуцирует у большинства пациентов повышение АД, хотя и разной степени выраженности. Авторы предположили, что причиной столь быстрого развития АГ может быть острая ингибиция VEGF-зависимой вазодилатации [35]. Точный механизм АГ при дефиците VEGF неизвестен, однако обсуждаются несколько причин АГ как следствия его блокады. Так, B. Li и соавт. удалось показать, что VEGFR-2 является основным медиатором гипотензивного эффекта VEGF, развивающегося в результате ва- зодилатации, обусловленной высвобождением из клеток эндотелия оксида азота и простациклина [27]. Таким образом, блокада VEGF независимо от её механизма - избыток sFlt-1 при ПЭ или медикаментозная VEGF-аблация - способна вызвать развитие АГ. Другой обсуждаемой причиной АГ при дефиците VEGF является уменьшение площади микроциркуляторного русла, что приводит к увеличению периферического сосудистого сопротивления и снижению активности оксида азота [32]. Независимо от этого VEGF оказывает гипотензивное действие, воздействуя на барорецепторы эндотелиоцитов [37], а его блокада противодействует этому. Кроме того, блокада VEGF нарушает его баланс с эндотелином, который является мощным вазоконстриктором [38]. Эти данные подтверждают результаты исследования О.В. Зозули и соавт., установивших, что дефицит оксида азота и простациклина вызывает развитие гипертензивных осложнений беременности и играет ключевую роль в формировании плацентарной недостаточности, тогда как избыток эндотелина и фибронектина, выявляемый только при гипертензивных осложнениях беременности (максимально - при ПЭ), коррелирует с тяжестью АГ и параметрами почечной функции [39].
Механизм развития протеинурии при ПЭ до недавнего времени оставался загадкой. Как и снижение СКФ, ПУ связывали с гломерулярным эндотелиозом, считавшимся патогномоничным морфологическим признаком ПЭ. Однако обнаружение признаков эндотелиоза у беременных с ге- стационной АГ без протеинурии и даже у здоровых беременных женщин заставило искать иное объяснение. Предпринятые в последние годы исследования позволили установить, что у пациенток с ПЭ обусловленный избытком sFlt-1 дефицит VEGF нарушает его аутокринную функцию в отношении подоцитов. Физиологическая концентрация VEGF необходима для сохранения гомеостаза и выживаемости подоцитов, а также поддержания функции щелевой диафрагмы за счет регуляции экспрессии нефрина. Повышенная концентрация sFlt-1 снижает экспрессию белков щелевой диафрагмы, в первую очередь, нефрина, что приводит к протеинурии [31, 32, 40, 41]. Важная роль подоцитов подтверждается и другими исследованиями. Так, V. Garovich и соавт. продемонстрировали снижение экспрессии нефрина и синаптопо- дина в клубочках почек женщин, умерших от ПЭ, и у экспериментальных животных при введении им sFlt-1 или анти-VEGF-антител [42]. Те же авторы были одними из первых, установивших наличие подоцитов в моче женщин с ПЭ. По данным V. Garovic и соавт., подоцитурия была выявлена у 15 из 15 женщин с ПЭ, отсутствовала - у 16 из 16 женщин с неосложненной беременностью, а также у 7 женщин с другими причинами АГ, ПУ или заболеваниями почек. При этом у тех пациенток, которым потом диагностировали ПЭ, подоциты в моче появлялись уже во II триместре беременности, давая основание считать подоцитурию ранним маркером ПЭ [43]. Позже аналогичные данные были получены другими авторами [44, 45]. При изучении подоцитурии, как маркера подоци- топатии, отмечены высокая специфичность и чувствительность показателя [45], что подтверждает предположение о роли подоцитарного повреждения в генезе преэкламптической протеинурии.
Таким образом, на сегодняшний день существует немало доказательств того, что ингибиция VEGF рецепторами sFlt-1 или анти-VEGF препаратами способна вызвать гломерулярное поражение. Применительно к ПЭ можно предполагать, что это поражение сочетает в себе гломерулярный эндотелиоз и подоцитопатию (рисунок).
В начале XXI века в ряде эпидемиологических исследований было убедительно продемонстрировано, что женщины, перенесшие ПЭ во время беременности, в последующем имеют высокий риск развития АГ, ишемической болезни сердца, острого инфаркта миокарда или инсульта, в связи с чем ПЭ была отнесена к факторам риска сердечно-сосудистых заболеваний (ССЗ) [46, 47]. Принимая во внимание сходство факторов риска сердечно-сосудистых осложнений и хронической болезни почек (ХБП), преэклампсию можно рассматривать также и как фактор риска ХБП.
В 2008 г. учеными из Норвегии было опубликовано крупное исследование, показавшее, что рождение «маловесных» детей сопряжено с риском развития в последующие годы ХБП у матерей [48]. Эта уникальная работа, проводившаяся в течение почти 40 лет, позволила проанализировать связь частоты развития терминальной почечной недостаточности и перенесенной ПЭ. Оказалось, что риск развития ХБП у женщин, перенесших преэклампсию, был в 4-5 раз выше, чем в популяции. В группе максимального риска оказались женщины с повторными ПЭ и женщины, родившие детей с низкой массой тела относительно гестационного срока.
Очевидно, что существуют несколько механизмов, связывающих ПЭ с последующей ХБП. К ним можно отнести АГ, сохраняющуюся у ряда пациенток, перенесших тяжелую ПЭ, эндотелиальную дисфункцию, ожирение [49]. Оказалось также, что после перенесенной ПЭ длительно сохраняется микроальбуминурия (МАУ). У 20-40% женщин, не имевших болезней почек до беременности, в течение нескольких лет после родоразрешения выявляли МАУ и повышенные цифры АД. Факт того, что у достаточно большого числа пациенток после ПЭ персистирует МАУ, указывает на возможность необратимого гломерулярного повреждения [50, 51]. Кроме того, протеинурия, в том числе и МАУ, сама по себе вызывает прогрессирующую почечную дисфункцию за счет усиления интерстициального воспаления [52]. По данным литературы, до 20% женщин, перенесших ПЭ, имеют признаки ХБП после родоразрешения [53, 54]. Между тем, механизмы хронизации почечного поражения при преэклампсии, особенно ее раннем развитии - до 34 нед беременности, практически неизвестны нефрологам, так как в современной литературе основной целью сравнения «ранней» и «поздней» ПЭ является изучение акушерских аспектов проблемы (состояние маточно-плацентарного кровотока и перинатальной смертности) [55, 56].
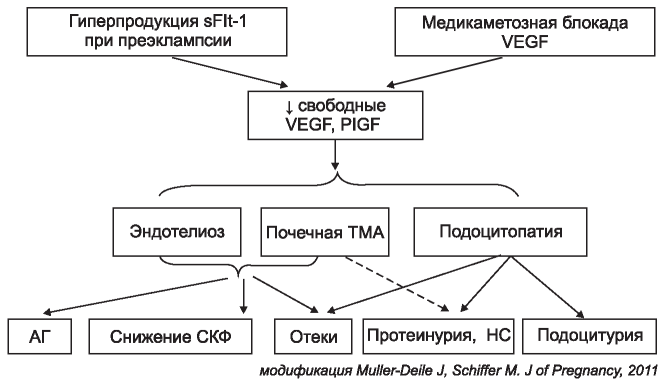
Блок-схема поражения почек при блокаде VEGF
В проведенном нами исследовании была поставлена задача изучить влияние ПЭ на формирование ХБП при «раннем» и «позднем» развитии осложнения. Оказалось, что у женщин, перенесших «позднюю» ПЭ, АД нормализовалось, а ПУ снизилась до минимальной уже на следующий день после родов, функция почек была нормальной. Эти параметры не изменились в течение следующих 12 мес. Напротив, после перенесенной «ранней» ПЭ в течение этого времени сохранялась АГ, требующая применения антигипертензивных препаратов, суточная ПУ составляла 0,5 г, СКФ - 71-74 мл/мин. Таким образом, у части пациенток уже в исходе ранней ПЭ формируется ХБП. При анализе мониторируемых показателей спустя 5 лет после родов АГ отмечалась в обеих группах пациенток, мочевой синдром был представлен небольшой изолированной ПУ, а СКФ соответствовала ХБП 2 ст. независимо от срока развития ПЭ. Полученные результаты свидетельствуют о том, что формирование ХБП происходит как после «ранней», так и после «поздней» ПЭ, однако в разные сроки [11, 12].
Клиническая картина нефропатии при «ранней» ПЭ отличается от «поздней». Избыточно высокий уровень sFlt-1 в группе пациенток с «ранним» её развитием подтверждает вклад ингибирования VEGF в формирование тяжелого поражения почек именно при «ранней» ПЭ (при «поздней» ПЭ уровень sFlt-1 не отличался от такового у здоровых беременных). Поэтому мы предположили, что «ранняя» и «поздняя» ПЭ - суть отдельные клинические ситуации, которые имеют не только различные акушерские исходы, но и различный «почечный» прогноз [11, 12].
ПЭ представляет собой патологию второй половины беременности, в основе которой лежит системная эндотелиальная дисфункция, обусловленная дисбалансом ангиогенных и антиангиогенных факторов, вырабатываемых плацентой. Основным проявлением «материнского синдрома» при ПЭ являются почки. В основе их поражения, представленного сочетанием гломерулярного эндоте- лиоза и повреждения подоцитов, лежит дефицит VEGF, обусловленный избыточным содержанием в циркуляции sFlt-1, блокирующим аутокринные и паракринные эффекты VEGF, направленные на поддержание структурно-функциональной целостности клубочков. Результатом его нарушения являются АГ, ПУ и снижение СКФ.
ПЭ является фактором риска ССЗ и ХБП в отдаленном будущем, а рождение «маловесных» детей сопряжено с развитием тПН у их матерей спустя многие годы после родов. Учитывая, что рано развившаяся ПЭ характеризуется более тяжелыми клиническими проявлениями и ассоциирована с неблагоприятным исходом как для матери, так и для плода, вероятно, ее следует рассматривать как тяжелое заболевание беременных женщин, первично поражающее плаценту и приобретающее системный характер вследствие генерализации поражения сосудистого эндотелия. «Поздняя» ПЭ, в отличие от «ранней», скорее, представляет собой синдром, клинические проявления которого выражены в значительно меньшей степени, несмотря на общность механизмов развития. Правомерность такого предположения обоснована недавними сведениями о том, что гломерулярный эндотелиоз - морфологический феномен, обусловленный беременностью как таковой, и при локальных формах не проявляет себя клинически, что подтверждается обнаружением его гистологических признаков в биоптатах почек не только у пациенток с индуцированной беременностью АГ, но и у здоровых женщин в III триместре беременности [18, 20]. С этой позиции ПЭ представляет собой крайне выраженное проявление эндотелиоза [18], а «ранняя» ПЭ, с нашей точки зрения, вероятно, может рассматриваться как его катастрофический вариант, характеризующийся тяжелым течением и неблагоприятным прогнозом для матери и плода.
1. James PR, Nelson-Piercy C. Management of hypertension before, during and after pregnancy. Heart 2004; 90: 1499–1504. doi.org/10.1136/hrt.2004.035444
2. Noris M, Perico N, Remuzzi G. Mechanisms of disease: pre-eclampsia. Nature Clin Pract Nephrol 2005; 1(2): 98–114. doi.org/10.1038/ncpneph0035
3. Нertig A, Watnick S, Strevens H. et al. How should women with pre-eclampsia be followed up? New insights from mechanistic studies. Nature Clin Pract Nephrol 2008; 4 (9): 503—509. doi.org/10.1038/ncpneph0880
4. Levine RJ, Maynard SE, Quan C et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 2004; 350: 672–683. doi.org/10.1056/NEJMoa031884
5. Maynard SE, Min JY, Lim KH et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt-1) may contribute to endothelian dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 2003; 111: 649–658. doi.org/10.1172/JCI17189
6. Karumanchi SA, Maynard SE, Stillman IE et al. Preeclampsia: a renal perspective. Kidney Intern 2005; 67: 2101–2113. doi.org/10.1111/j.1523-1755.2005.00316.x
7. Phyllis A. Preeclampsia: a «nephrocentric» view. Adv Chronic Kidney Rev 2013; 20(3):280–286 doi.org/10.1053/j.ackd.2013.01.013
8. Тареева ИЕ. Нефрология: Руководство для врачей. Медицина, М., 2000; 464–473 [Tareeva I.E. Rukovodstvo dlya vrachej. Medicina, M., 2000; 464–473]
9. Fisher KA, Ahuja S, Luger AM et al. Nephrotic proteinuria with preeclampsia. Am J Obstet Gynecol 1977; 129: 643–646. doi.org/10.1016/0002-9378(77)90646-9
10. Cohen AW, Burton NG. Nephrotic syndrome due to preeclamptic nephropathy in hydatidiform mole and coexistent fetus. Obstet Gynaecol 1979; 53: 130–134
11. Козловская НЛ, Меркушева ЛИ, Кирсанова ТВ и др. Влияние дисбаланса плацентарных факторов ангиогенеза на клинические проявления «ранней» и «своевременной» преэклампсии. Нефрология и диализ 2013; 15(3): 206–215 [Kozlovskaya NL, Merkusheva LI, Kirsanova TV i dr. Vliyanie disbalansa platsentarnykh faktorov angiogeneza na klinicheskie proyavleniya «rannej» i «svoevremennoj» preehklampsii. Nephrology and Dialysis 2013; 15(3):206–215]
12. Козловская НЛ, Меркушева ЛИ, Кирсанова ТВ и др. Дисбаланс плацентарных факторов ангиогенеза и клинические особенности «ранней» и «своевременной» преэклампсии. Взгляд нефролога. Архив акушерства и гинекологии им. В.Ф.Снегирева 2014; 1:13–21 [Kozlovskaya NL, Merkusheva LI, Kirsanova TV i dr. Disbalans platsentarnykh faktorov angiogeneza i klinicheskie osobennosti «rannej» i «svoevremennoj» preehklampsii. Vzglyad nefrologa. V.F.Snegirev Archives of Obstetrics and Gynecology 2014; 1:13–21]
13. Davison JM, Dunlop W. Renal hemodynamics and tubular function normal human pregnancy. Kidney Int 1980; 18: 152–161. doi.org/10.1038/ki.1980.124
14. Moran P, Baylis PH, Lindheimer MD. Glomerular ultrafiltration in normal and preeclamptic pregnancy. J Am Soc Nephrol 2003;14: 648–652. doi.org/10.1097/01.ASN.0000051724.66235.E0
15. National Kidney Foundation. Kidney Disease Outcome Quality Initiative (KDOQI). Clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Am J Kidney Dis 2007; 49 (2): 1–180. DOI: 10.1053/j. ajkd.2006.12.005
16. Smith МС, Moran Р, Ward MK, Davison JM. Assessment of glomerular filtration rate during pregnancy using the MDRD formula. BJOG: An International Journal of Obstetrics & Gynaecology 2008; 115(1): 109–112. doi.org/10.1111/j.1471-0528.2007.01529.x
17. Schaffer N, Dill L, Cadden J. Uric acid clearance in normal pregnancy and preeclampsia. J Clin Invest 1943; 22:201–206. doi.org/10.1172/JCI101383
18. Stillman IE, Karumanchi SA. The glomerular injury of preeclampsia. J Am Soc Nephrol 2007;18:2281–2284. doi.org/10.1681/ASN.2007020255
19. Baumwell S, Karumanchi SA. Pre-eclampsia: clinical manifestation and molecular mechanisms. Nephron Clin Pract 2007;106:72–81. doi.org/10.1159/000101801
20. Strevens H, Wide-Swensson D, Hansen A еt al. Glomerular endotheliosis in normal pregnancy and pre-eclampsia. Br J Obst Gynecol 2003;110: 831–836. doi.org/10.1111/j.1471-0528-.2003.02162.x
21. Gaber LW, Spargo BH: Pregnancy-induced nephropathy: The significance of focal segmental glomerulosclerosis. Am J Kidney Dis 1987; 9: 317–323. doi.org/10.1016/S0272-6386-(87)80129-4
22. Heaton JM, Turner DR. Persistent renal damage following pre-eclampsia: A renal biopsy study of 13 patients. J Pathol1985; 147: 121–126. doi.org/10.1002/path.1711470207
23. Saritas T, Moeller MJ. Pre-eclamsia, podocyturia and the role of parietal epithelial cells. J Nat Rev Nephrol 2014;10:615– 616.doi:10.1038/nrneph.2014.163
24. Simon M, Grone H-J, Johren O et al. Expression of vascular endothelial growth factor and its receptors in human renal ontogenesis and in adult kidney. Am J Physiol 1995; 37:24–250. doi.org/10.1152/ajprenal.1995.268.2.F240
25. Nakagawa T, Lan HY, Zhu HJ et al. Differential regulation of VEGF by TGF-beta and hypoxia in rat proximal tubular cells. Am J Physiol Renal Physiol 2004; 287(4): 658–664. doi.org/10.1152/ajprenal.00040.2004
26. Kearney JB, Kappas NC, Ellerstrom C et al. The VEGF receptor flt-1 (VEGFR-1) is a positive modulator of vascular sprout formation and branching morphogenesis. Blood 2004; 103(12): 4527 – 4535. doi.org/10.1182/blood-2003-07-2315
27. Li B, Ogasawara AK, Yang R et al. KDR (VEGF receptor 2) is the major mediator for the hypotensive effect of VEGF. Hypertension 2002;39(6):1095–1100. doi.org/10.1161/01.HYP.0000018588.56950.7A
28. Suga S, Kim Y, Joly A et al. Vascular endothelial grown factor (VEGF121) protects rats from renal infarction in thrombotic microangiopathy. Kidney International 2001;60: 1297–1308. doi.org/10.1046/j.1523-1755.2001.00935.x
29. Eremina V, Jefferson A, Kowalewska J et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 2008; 358: 1129–1139. doi.org/10.1056/NEJMoa0707330
30. Bollee, G, Patey N, Cazajous G et al. Thrombotic microangiopathy secondary to VEGF pathway inhibition by sunitinib. Nephrol Dial Transplant 2008; 24: 682–685. doi.org/10.1093/ndt/gfn657 31.
31. Ballermann BJ. Glomerular endothelial cell differentiation. Kidney Int 2005; 67:1668–1671. doi.org/10.1111/j.1523-1755-.2005.00260.x
32. Muller-Deile J, Schiffer M. Renal involvement in preeclampsia: similarities to VEGF ablation therapy. J of Pregnancy 2011; 2011: 6 pages. Article ID 176973. doi.org/10.1155/2011/176973
33. Duley L. The global impact of pre-eclampsia and eclampsia. Seminars in Perinatology 2009;33(3):130–137. doi. org/10.1053/j.semperi.2009.02.010
34. Yang JC, Haworth L, Sherry RM et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427–434. doi. org/10.1056/NEJMoa021491
35. Robinson E, Matulonis U, Ivy P et al. Rapid development of hypertension and proteinuria with cediranib, an oral vascular endothelial grown factor receptor inhibitor. Clinical Journal of the American Society of Nephrology 2010; 5(3): 477–483. doi. org/10.2215/CJN.08111109
36. Patel TV, Morgan GA, Demetri GD et al. A preeclampsialike syndrome characterized by reversible hypertension and proteinuria indused by the multitargeted kinase inhibitors sunitinib and soferenib. Journal of the National Cancer Institute 2008;100 (4):282–284. doi.org/10.1093/jnci/djm311
37. Yang R, Ogasawara AK, Zioncheck TF et al. Exaggerated hypotensive effect of vascular endothelial growth factor in spontaneously hypertensive rats. Hypertension 2002;39(3):815–820. doi.org/10.1161/hy0302.105398
38. Okuda Y, Tsurumaru K, Suzuki S et al. Hypoxia and endothelin-1 induce VEGF production in human vascular smooth muscle cells. Life Sciences 1998; 63(6):477–484.doi.org/10.1016/S0024-3205(98)00296-3
39. Зозуля ОВ, Рогов ВА, Пятакова НВ, Тареева ИЕ. Оксид азота: роль в развитии осложнений беременности и в их профилактике у женщин с гипертонической болезнью и хроническим гломерулонефритом. Терапевтический архив 1997; 69(6):17–21 [Zozulya OV, Rogov VA, Pyatakova NV, Tareeva IE. Oksid azota: rol' v razvitii oslozhnenij beremennosti i v ih profilaktike u zhenshchin s gipertonicheskoj bolezn'yu i hronicheskim glomerulonefritom. Terapevticheskij arhiv 1997;69(6):17–21]
40. Sugimoto H, Hamanog Y, Charytan D et al. Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF antibodies and soluble VEGF receptor 1 (sFlt-1) induces proteinuria. Journal of Biological Chemistry 2003;278(15):12605–12608. doi.org/10.1074/jbc.C300012200
41. Henao DE, Mathieson PW, Saleem MA et al. A novel renal perspective of preeclamsia: a look from the podocyte. Nephrol Dial Transplant 2007;22(5): 1477 .doi:10.1093/ndt/gf1804
42. Garovic V, Wagner S, Petrovic L et al. Glomerular expression of nephrin and synaptopodin, but not podocin, is decreased in kidney sections from women with preeclampsia. Nephrol Dial Transplant 2007;22: 1136–1143. doi.org/10.1093/ndt/gfl711
43. Garovic VD, Wagner SJ, Turner ST. Urinary podocyte excretion as a marker for preeclampsia. Am J Obst Gynecol 2007; 196: 320–327. dx.doi.org/10.1016/j.ajog.2007.02.007
44. Aita K, Etoh M, Hamada H et al. Acute and transient podocyte loss and proteinuria in preeclampsia. Nephron Clin Practice 2009; 112: 65–70. doi: 10.1159/000213083
45. Craici I M, Steven J. Wagner, Kent R. Bailey at all. Podocyturia Predates Proteinuria and Clinical Features of Preeclampsia. Hypertension 2013;61:6 1289–1296. doi.org/10.1161/HYPERTENSIONAHA.113.01115
46. Bellamy L, Casas JP, Hingorani AD, Williams DJ. Preeclampsia and risk of cardiovascular disease and cancer in later life: systematic review and meta-analysis. BMJ 2007; 335:974. doi.org/10.1136/bmj.39335.385301.BE
47. Rodie V, Freeman DJ, Sattar N et al. Pre-eclampsia and cardiovascular disease: metabolic syndrome of pregnancy. Atherosclerosis 2004; 175(2): 189–202. doi.org/10.1016/j.atherosclerosis.2004.01.038
48. Vikse В, Lorentz M. Irgens et al. Preeclampsia and the Risk of End-Stage Renal Diseasе. Engl J Med 2008; 359: 800–809. doi.org/10.1056/NEJMoa0706790
49. Munkhaugen J, Vikse BE. New aspects of pre-eclamsia: lessons for the nephrologist. Nephrol Dial Transplant 2009; 3 pages.doi:10.1093/ndt/gfp341
50. Bar J, Kaplan B, Wittenberg C et al. Microalbuminuria after pregnancy complicated by preeclampsia. Nephrol Dial Transplant 1999; 14: 1129–1132. doi.org/10.1093/ndt/14.5.1129
51. Nisell H, Lintu H, Lunell NO et al. Blood pressure and renal function seven years after pregnancy complicated by hypertension. Br J Obstet Gynaecol 1995;102: 876–881. doi.org/10.1111/j.1471-0528.1995.tb10874.x
52. Abbate M, Zoja C, Rumuzzi G. How does proteinuria cause progressive renal damage? J Am Sos Nephrol 2006; 17: 2974–2984. doi.org/10.1681/ASN.2006040377
53. Murakami S, Saitih M, Kubo T et al. Renal desease in woman with severe preeclampsia or gestational proteinuria. Obstet Gynaecol 2000; 96: 945–949. doi.org/10.1016/S00297844(00)01055-3
54. Reiter L, Brown MA, Whitworth JA. Hypertension in pregnancy: the incidence of underlying renal disease and essential hypertension. Am J Kidney Dis 1994; 24: 883–887. doi.org/10.1016/ S0272-6386(12)81055-9
55. Ihle BU, Long P, Oats J. Early onset preeclampsia: Recognition of underlying renal disease. Br Med J (Clin Res Ed)1987; 294:79. DOI: 10.1016/0020-7292(87)90109-3
56. Poon LCY, Kametas NA, Maiz N et al. First – trimestre prediction of hypertensive disorders in pregnancy. Hypertension 2009; 53: 812–818. doi: 10.1161/HYPERTENSIONAHA.108.127977
Меркушева Людмила Игоревна, кандидат медицинских наук, врач-нефролог
117197, Москва, ул. Академика Опарина, д. 4.
Козловская Наталья Львовна - профессор, доктор медицинских наук, кафедра внутренних, профессиональных болезней и пульмонологии медико-профилактического факультета
119991, Москва, ул. Трубецкая, д. 8, стр. 2.
Меркушева Л.И., Козловская Н.Л. ПОРАЖЕНИЕ ПОЧЕК ПРИ ПРЕЭКЛАМПСИИ: ВЗГЛЯД НЕФРОЛОГА (ОБЗОР ЛИТЕРАТУРЫ). Нефрология. 2018;22(2):30-38. https://doi.org/10.24884/1561-6274-2018-22-2-30-38
Merkusheva L.I., Kozlovskaya N.L. RENAL INJURY IN PREECLAMPSIA: THE VIEW OF NEPHROLOGIST (LITERATURE REVIEW). Nephrology (Saint-Petersburg). 2018;22(2):30-38. (In Russ.) https://doi.org/10.24884/1561-6274-2018-22-2-30-38
![]()

197101, Санкт-Петербург, улица Льва Толстого, дом 17, литер А,
редакция журнала «Нефрология»
e-mail: journal@nephrolog.ru



































